Kardiomyopathien
Definition
Erkrankung der Herzmuskulatur mit struktureller und funktioneller Einschränkung – vorausgesetzt eine erklärende KHK, Hypertonie, valvuläre oder angeborene Herzerkrankung wurde ausgeschlossen.
Einteilung in Phänotypen und ätiologische Beispiele
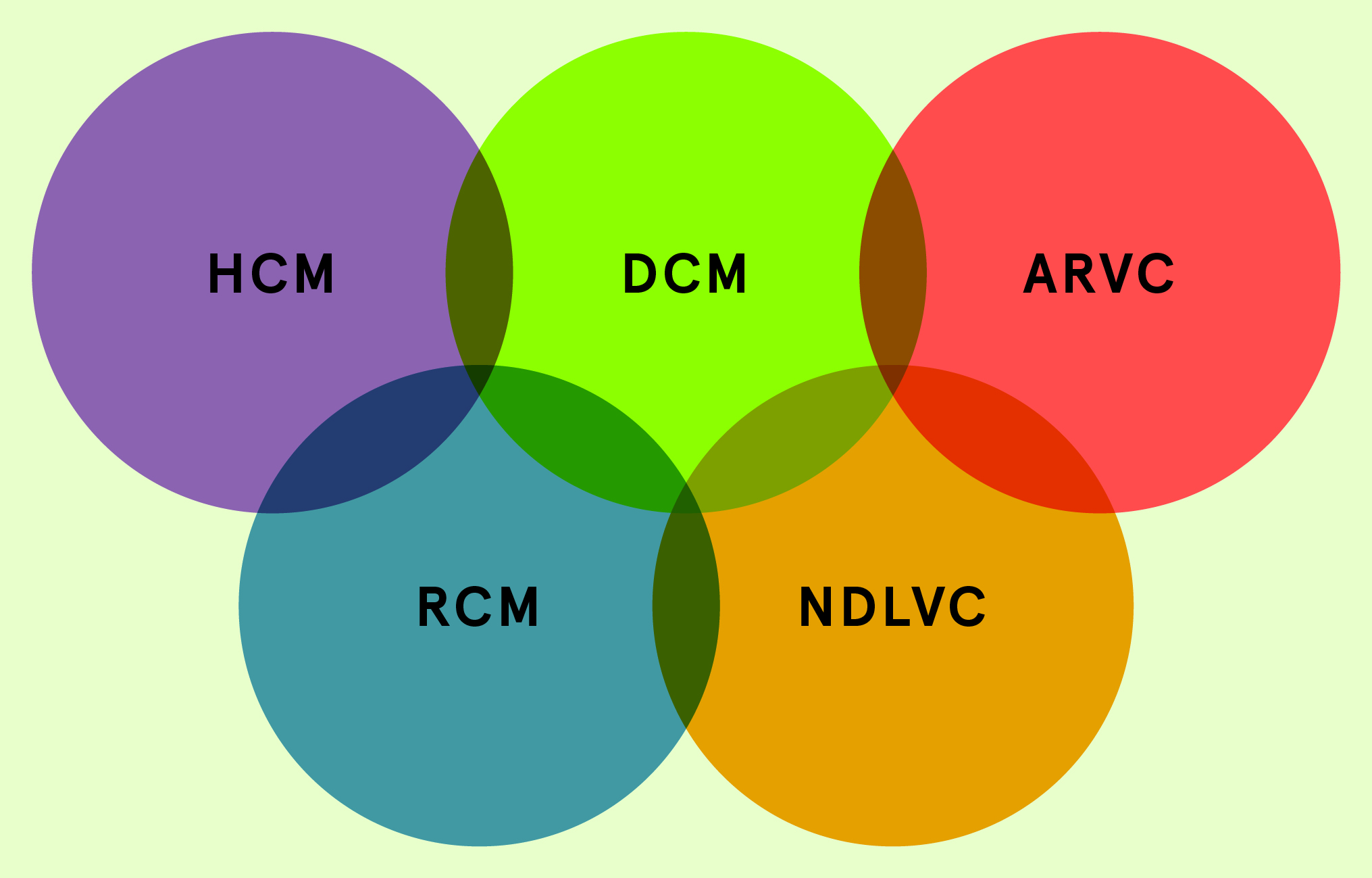
- Hypertrophe Kardiomyopathie (HCM): hypertrophe nicht-/obstruktive Kardiomyopathie im engeren Sinn (Sarkomererkrankung, s. unten ), aber auch (in der Regel mit Beteiligung des rechten Ventrikels) Speichererkrankungen wie z. B. kardiale Amyloidose , M. Fabry, Glykogenspeichererkrankungen etc.
- Dilatative Kardiomyopathie (DCM): Dilatation des linken Ventrikels mit systolischer Dysfunktion; gerade bei familiärer Häufung genetische Ursache suchen
- Nicht-dilatative linksventrikuläre Kardiomyopathie (NDLVC): im MRI nicht-ischämisches late gadolinium enhancement (LGE), häufig einer «Narbe» entsprechend; zu diesem Phänotyp gehört auch die linksdominante Form einer ARVC oder die kardiale Sarkoidose
- Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC): dominierende Dilatation des rechten Ventrikels und/ oder Dysfunktion, bei Vorliegen von gewissen histologischen, genetischen und/ oder elektrokardiographischen Kriterien
- Restriktive Kardiomyopathie (RCM): restriktives Füllungsmuster eines/ beider Ventrikel im Vordergrund
Diagnostik allgemein
Im Vordergrund stehen hier neben einer ausführlichen Anamnese (kardiale, bewusst aber auch extrakardiale Systemanamnese; Familienanamnese über 3 Generationen) das EKG sowie bildgebende Verfahren wie zunächst die Echokardiographie, fast immer aber auch ein kardiales MRI (IB-Indikation). Mit der Phänotypisierung beginnt erst der eigentliche Diagnostikprozess. Gerade aber auch bei syndromalen und metabolischen Erkrankungen mit kardialer Beteiligung (z. B. Morbus Fabry, RASopathien wie Noonan-Syndrom, neuromuskulären Erkrankungen, Mitochondriopathien, Glykogenspeichererkrankungen etc.) ist häufig die extrakardiale Anamnese richtungsweisender als der Phänotyp.
Für eine genetische Beratung und dann ggf. auch Abklärung besteht für fast alle Kardiomyopathien formal eine Klasse IB-Indikation. Aufgrund der deutlich eingeschränkten Ressourcen, aber auch potentieller Verunsicherung durch inkonklusive Befunde muss dies aber im Einzelfall entschieden werden.
Grundsätzlich gilt: umso jünger die Patientin oder der Patient, umso mehr Organsysteme betroffen sind, umso auffälliger die Befunde – desto intensiver soll eine spezifische Ätiologie gesucht werden.
Häufige, spezifische Kardiomyopathien
Hypertrophe Kardiomyopathie (HCM)
- Im engeren Sinne (i.d.R. genetische) Erkrankung des Sarkomers
- Wesentlicher Grund für die Belastungsabhängigkeit klinischer Beschwerden ist die dynamische Obstruktion des linksventrikulären Ausflusstrakts (LVOT), die bei Volumenmangel und Hyperdynamie des linken Ventrikels zunimmt, und häufig dann auch eine strukturelle Mitralklappeninsuffizienz verstärkt; dies besteht bei 1/3 der Patientinnen und Patienten bereits in Ruhe, bei 1/3 nur unter Belastung (oder Valsalvamanöver); bei 1/3 der Patientinnen und Patienten findet sich keine Obstruktion (HNOCM, im Ggs. zur HOCM)
Epidemiologie
Prävalenz 0.2-0.5%, aber nur 15% klinisch identifiziert
Klinik
Sehr variabel und wenig spezifisch
- Supra- (v.a. VHFlimmern) und ventrikuläre Rhythmusstörungen/ plötzlicher Herztod
- Beschwerden i.d.R. belastungsabhängig wie Dyspnoe, Angina pectoris, Schwindel, Prä-/Synkopen; oft starke Tagesform-Abhängigkeit (Wärme, postprandial etc.)
Diagnostik (neben v.a. auch Familien-Anamnese)
- EKG: Zeichen der linksventrikulären Hypertrophie, je nach Phänotyp häufig sehr ausgedehnte Repolarisationsstörungen (unauffälliges EKG nur in 5-10%)
- Echokardiographie: meist asymmetrische Hypertrophie meist des linken Ventrikels, Papillarmuskel- und Mitralklappenpathologien
- MRI: neben Darstellung der Anatomie bessere Gewebecharakterisierung, z.B. diffuse Vernarbung (late gadolineum enhancement/LGE) v.a. im Bereich der hypertrophen Areale, T1-Mapping etc.)
- Genetik: in 30-40% wahrscheinlich/ pathogene Mutation detektierbar
Risikostratifizierung
- ESC HCM-Risk calculator (Alter, Wanddicke, Vorhof-Grösse, LVOT-Gradient, pos. FA für plötzlichen Herztod, NSVT, Synkopen)
- Ausmass LGE
Therapie
- Symptomatisch: v.a. durch Reduktion der LVOT-Obstruktion (primär durch Vermeiden einer Volumendepletion – keine Diuretika!)
- Medikamentös: primär Versuch mit negativen Inotropika wie BBlocker oder Nichtdihydropyridin-Calcium-Antagonisten (Diltiazem, Verapamil), sekundär Myosininhibition durch Mavacamten
- Interventionell/chirurgisch: transkoronare Ablation der Septumhypertrophie (TASH), Myektomie
- Prophylaktisch: ICD
Kardiale Amyloidose
Verschiedene Proteine (bekannt mind. 42) können zur Bildung unlöslicher Amyloidfibrillen führen, die dann extrazellulär abgelagert werden (u.a. im Herz); mit Abstand am häufigsten ist hier das Transthyretin- (ATTR), sehr viel seltener das AL-Amyloid (monoklonale Immunglobulin-Leichtketten, hämatologische Erkrankung)
Epidemiologie
Initial als «rare/orphan disease» interpretiert, geht man heutzutage von einer nicht seltenen Erkrankung aus (Nachweis von ATTR-Amyloid in 25% aller Autopsien > 80-Jähriger, 12% bei HFpEF-, ca. 10% bei schweren Aortenstenosepatientinnen und -patienten). Jährlich zeigt sich eine stark steigende Inzidenz aufgrund zunehmender Awareness. Am häufigsten ist die Wildtyp-Variante (> 90-95 %; meist Männer im 7. oder 8. Lebensjahrzehnt), die hereditäre Variante ist (vor allem in unseren Breitengraden) sehr selten (Betroffene in aller Regel deutlich jünger, häufig auch neurologischer Phänotyp im Vordergrund).
Klinik
- Kardial: Herzinsuffizienz, Vorhofflimmern, Reizleitungsstörungen mit Schrittmacherbedürftigkeit
- Extrakardial: muskuloskelettal (Carpaltunnelsyndrom v.a. auch bds. sehr häufig, Spinalkanalstenose, Bizeps-/Sehnenrupturen, Spickfinger, Schmerzen der grossen Gelenke), Polyneuropathie, autonome Dysfunktion (orthostatische Hypotonie, chronische Diarrhoe/Obstipation, erektile Dysfunktion)
Diagnostik
- EKG: im Ggs. zu den hypertroph wirkenden Ventrikeln niedrige QRS-Amplituden (eher selten «echte» periphere low voltage, häufiger «pseudoinfarction pattern» in V1-4)
- Echokardiographie: biventrikuläre Hypertrophie mit im Vordergrund stehender diastolischer Dysfunktion und (sehr sensitiv, aber wenig spezifisch) klassischem Strainmuster, biatriale Dilatation, Perikarderguss, Verdickung von AV-Klappen und interatrialem Septum
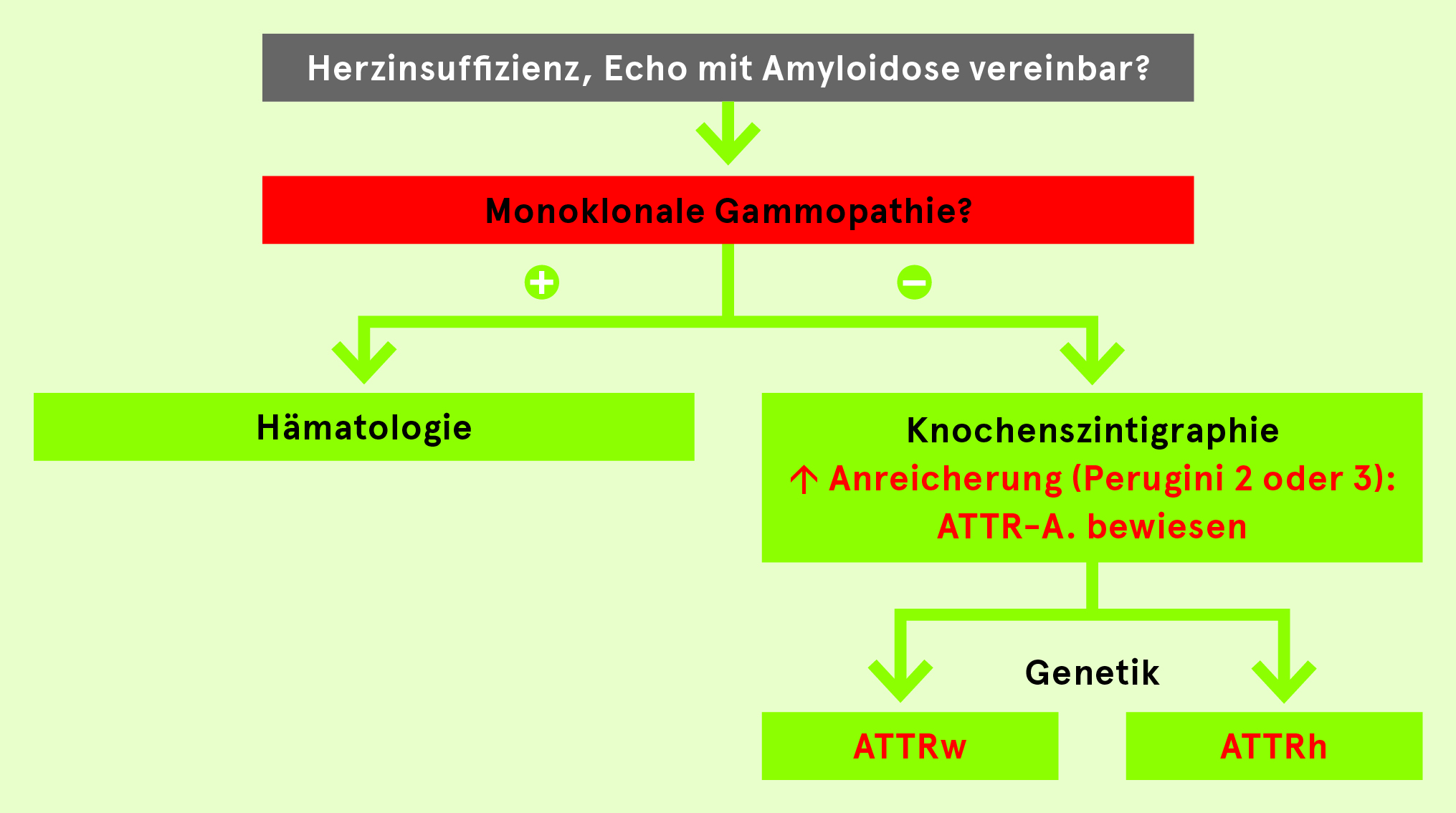
- Labor: neben oft sehr hohem NT-proBNP und erhöhtem Troponin v.a. Ausschluss AL-Amyloidose (Immunelektrophorese im Serum und im Urin, freie Leichtketten im Serum; bei Unsicherheit (hohe Prävalenz eines MGUS!) Rücksprache mit der Hämatologie
- Knochen-Szintigraphie: deutliche Anreicherung myokardial, sehr hohe Sensitivität, aber auch Spezifität
- MRI: klassische Befunde, meist aber nicht notwendig für die Diagnosestellung
- Biopsien: meist nicht notwendig – bei diagnostischer Unsicherheit myokardial (Bauchfett/Rektum/Unterlippen-Biopsien i.d.R. negativ!)
- Genetik: v.a. bei jüngeren Patientinnen und Patienten isoliert Untersuchung des TTR-Gens
Therapie
Überleben wird statistisch verlängert allein schon durch höhere Awareness und dadurch frühere Diagnose.
- Klassische Herzinsuffizienzmedikation: Grundsätzlich ja, Datenlage aber ungenügend, und häufig schlecht toleriert (im Fokus a.e. MRAs und SGLT2-A.)
- Spezifische Therapie: grundsätzlich 3 Angriffspunkte (Suppression TTR-Synthese, TTR-Stabilisierung, Abbau bereits abgelagerter Amyloidfibrillen); zugelassen bisher nur der TTR-Stabilisator Tafamidis mit sehr guter Datenlage und Verträglichkeit (aufgrund hohem Preis Kostengutsprachengesuch durch spezialisierte Zentren wie das KSSG notwendig); interessante alternative/zusätzliche Substanzen in allen 3 Angriffspunkten in der Entwicklung/klinischen Studien
Kardiale Sarkoidose
- Insgesamt noch viele Unklarheiten hinsichtlich Pathogenese (Kombination aus genetischen und Umwelt-Faktoren), Epidemiologie, Risikostratifizierung und therapeutischem Management
- Wichtige Differenzialdiagnosen: Desmoplakin-Kardiomyopathie, ARVC
- Screening auf kardialen Befall bei allen Sarkoidosepatientinnen und -patienten mit Anamnese, EKG, 24h-Holter und Echokardiographie
- Regelmässige Diskussion am Sarkoidoseboard (Pneumologie, Radiologie, Pathologie, Rheumatologie, Neurologie, Nephrologie, Kardiologie, ggf. auch Ophtalmologie und Dermatologie)
Epidemiologie
Kardiale Beteiligung bei systemischer Sarkoidose um 25-30% (Autopsie-respektive MRI-Untersuchungen; klinisch evident nur in 5-10%); Prävalenz isolierter kardialer Sarkoidose 3-9%; starke regionale Unterschiede (Nord-Süd-Gefälle); deutliche Zunahme der Diagnosestellung in den letzten 2 Jahrzehnten (zugenommene Awareness und grosszügigere Bildgebung)
Klinik
Höhergradiger AV-Block gerade bei jungen Patientinnen und Patienten, Herzinsuffizienz, ventrikuläre Rhythmusstörungen
Diagnostik (für Erstdiagnose, Risikostratifizierung wie auch Verlauf)
- Kardiale Biomarker (Troponin, natriuretische Peptide)
- EKG: AV-Blockierungen/ Schenkelblockbilder/ ventrikuläre Rhythmusstörungen
- Kardiale Bildgebung: Echokardiographie, v.a. aber MRI und 18F-FDG-PET (entsprechende Ernährungs-Vorbereitung mit Suppression des Glukose-Stoffwechsels essentiell)
- Histologie/Biopsie: je nach angewandten Diagnosekriterien essentiell, extrakardial oder endomyokardial (Problem: oft falsch-negativ aufgrund fokalem Befall)
Therapie
- Devices: i.d.R. aufgrund AV-Blockierung Schrittmacher, häufig als kardiale Resynchronisationstherapie (CRT) und je nach Risikostratifizierung meist inklusive Defibrillator zur Primärprophylaxe maligner Rhythmusstörungen
- Klassische Herzinsuffizienz-Medikation
- Immunsuppression: Start mit Steroiden, i.d.R. bereits initial Kombination mit Methotrexat oder Azathioprin; im Verlauf ggf. TNFa-Blocker
Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)
- Aufgrund nicht selten rein oder zumindest betont linksventrikulärer Beteiligung auch «arrhythmogene Kardiomyopathie» genannt
- Genetische Erkrankung mit Mutationen v.a. desmosomaler Proteine; Prävalenz 1:2000-1:5000
Klinik
Synkope, (supra- und) ventrikuläre Rhythmusstörungen, SCD
Diagnostik (revidierte Taskforce Kriterien von 2010, resp. Padua-Kriterien von 2020)
- Familienanamnese
- EKG (T-Negativierungen V1-4, Epsilon-Welle), Holter-EKG
- Echokardiographie/MRI (Volumen und EF des RV, regionale Kinetik)
- Genetik (positiv in ca. 2/3, 20-46% PKP2)
Risikostratifizierung
- SCD, VTs, Alter, männliches Geschlecht, Synkope, Ausdehnung T-Negativierungen, VES-Last, Einschränkung RVEF/LVEF
- www.arvcrisk.com
Therapie
- Sportrestriktion
- Medikamentös (BBlo, ggf. Flecainid)
- ICD
- Ablation bei rezidivierenden VTs
Quellen/Links
Arbelo E et al. 2023 ESC Guidelines for the management of cardiomyopathies. European Heart J. 2023, 44. https://www.escardio.org/Guidelines/Clinical-Practice-Guidelines/Cardiomyopathy-Guidelines
Dr. Niklas Ehl