Pulmonale Hypertonie
Definition
Die pulmonale Hypertonie (PH) ist neu definiert als Erhöhung des mittleren pulmonal-arteriellen Drucks (=Druck in der Arteria pulmonalis; mPAP) > 20 mmHg im Rechtsherzkatheter in Ruhe (ESC/ERS Leitlinien 2022). Die hämodynamische Messung im Rechtsherzkatheter ist der Gold-Standard und kann für eine definitive Diagnose und hämodynamische Klassifikation einer PH nicht durch die Echokardiografie ersetzt werden. Die pulmonal-arterielle Hypertonie (PAH) ist kein Synonym für PH sondern eine Untergruppe der PH (vgl. unten).
Häufigkeit
Man geht heute davon aus, dass 1% der Weltbevölkerung von einer PH betroffen ist. Die häufigste Form der PH ist die PH bei zugrundeliegender Linksherzerkrankung (Gruppe 2), gefolgt von der PH bei Lungenerkrankung (Gruppe 3). Diese beiden Gruppen machen zusammen > 80% der Fälle einer PH aus. Die PAH dagegen ist eine seltene Krankheit. Die Inzidenz der PAH beträgt in Europa etwa 5–10 auf 1’000’000/Jahr und die Prävalenz etwa 15–60 auf 1’000’000. Die PAH gilt deshalb als «orphan disease». Frauen sind häufiger betroffen. Am häufigsten ist dabei die idiopathische PAH. Unter den Formen der assoziierten PAH sind die Kollagenosen am häufigsten vertreten, insbesondere die systemische Sklerose.
Hämodynamische Definitionen der pulmonalen Hypertonie im Rechtsherzkatheter
| Definition | Hämodynamische Kriterien* | Klinische Gruppe* |
| Pulmonale Hypertonie (PH) | mPAP > 20 mmHg (neu, vorher ≥ 25 mmHg) |
Alle |
| Prä-kapilläre PH | mPAP > 20 mmHg und
mPAWP ≤ 15 mmHg und neu (2022) obligat: PVR ≥ 3 WU |
Gruppe 1, 3,4, 5 |
| Post-kapilläre PH | mPAP > 20 mmHg
mPAWP > 15 mmHg; immer Differenzierung in isoliert post-kapilläre PH (IpcPH) oder kombiniert prä- und post-kapilläre PH (CpcPH) |
Gruppe 2 (5) |
| – IpcPH | und PVR < 3 WU (neu kein Kriterium mehr für den diastolischen Druckgradienten) | Gruppe 2 |
| – CpcPH | und PVR ≥ 3 WU (neu kein Kriterium mehr für den diastolischen Druckgradienten) | Gruppe 2 (theoretisch können auch zwei Entitäten vorliegen: eine post-kapilläre PH und eine unabhängige prä-kapilläre PH) |
| Unklassifizierte PH (neu 2022) | mPAWP ≤ 15 mmHg und PVR < 3 WU d. h. erhöhter pulmonaler Fluss |
keine |
| PH unter Belastung (neu 2022) | Steigung mPAP/cardiac output von Ruhe zu Belastung >3 mmHg/l/min | keine |
Klassifikation der Pulmonale Hypertonie
Die PH wird gemäss ihrer Ätiologie in fünf klinische Gruppen eingeteilt. Die PAH ist die Bezeichnung für die Gruppe 1 (und kein Synonym für PH). Im Gegensatz dazu versteht man unter Funktionsklassen eine Einteilung des klinischen Schweregrads (analog NYHA-Klasse), welche primär für PatientInnen mit PAH angewendet wird (Unterkapitel Symptome der PAH Seite).
Klassifikation (5 klinische Gruppen)
| 1. Pulmonal-arterielle Hypertonie (PAH) | 1.1. Idiopathisch 1.1.1. Non-responder auf Vasoreaktivitätstestung 1.1.2. Akute Responder auf Vasoreaktivitätstestung 1.2. Hereditär 1.3. Arzneimittel- oder Toxin-induziert 1.4. Assoziiert mit: 1.4.1. Kollagenosen 1.4.2. HIV-Infektion 1.4.3. Portale Hypertension 1.4.3. Kongenitalen Herzkrankheiten 1.4.5. Schistosomiasis 1.5. PAH mit venöser/kapillärer Beteiligung 1.6. Persistierende PH des Neugeborenen |
| 2. PH assoziiert mit Linksherzerkrankungen | 2.1. Herzinsuffizienz 2.1.1. Herzinsuffizienz mit erhaltener Auswurffraktion 2.1.2. Herzinsuffizienz mit reduzierter oder leicht reduzierter Auswurffraktion 2.2. Klappenerkrankungen 2.3. Kongenitale/erworbene kardiovaskuläre Erkrankungen mit post-kapillärer PH |
| 3. PH assoziiert mit Lungenerkrankungen und/oder Hypoxie | 3.1. Obstruktive Lungenerkrankungen/Emphysem
3.2. Restriktive Lungenerkrankung 3.3. Lungenerkrankungen mit gemischt obstruktiv/restriktivem Muster 3.4. Hypoventilations-Syndrome 3.5. Hypoxie ohne Lungenerkrankung (z.B. Höhe) 3.6. Entwicklungsstörungen der Lunge |
| 4. PH assoziiert mit Pulmonalarterienobstruktion | 4.1. Chronisch thromboembolische PH (CTEPH) 4.2. Anderen Pulmonalarterienobstruktionen (z.B. Tumor) |
| 5. PH mit unklarem/multifaktoriellem Mechanismus | 5.1. Hämatologische Krankheiten 5.2. Systemerkrankungen 5.3. Metabolische Störungen 5.4. Chronische Niereninsuffizienz (mit/ohne Dialyse) 5.5. Pulmonale thrombotische Tumormikroangiopathie 5.6. Fibrosierende Mediastinitis |
Anamnese
- Pulmonale/kardiale Vorerkrankungen
- Familiäre Häufung
- Rheumatologische Grunderkrankung (systemische Sklerose, systemischer Lupus erythematodes [SLE] , rheumatoide Arthritis [RA])
- Lebererkrankungen, HIV
- Lungenembolien/Thrombosen
- Medikamente/Drogen (siehe folgende Tab.)
Medikamente, Drogen und Toxine assoziiert mit PAH
| Gesichert | Wahrscheinlich | |
| Aminorex | Kokain | Interferon-α und -β |
| Dexfenfluramin | Phenylpropanolamin | Alkylierende Substanzen |
| Fenfluramin | L-Tryptophan | Bosutinib |
| Benfluorex | Hypericum perforatum
(echtes Johanniskraut) |
Leflunomid |
| Toxisches Rapsöl | Diazoxid | Indirubin |
| Metamphetamine | Amphetamin | Ponatinib |
| Dasatinib | Sofosbuvir | Carfilzomib |
Symptome der Pulmonalen Hypertonie
Die Symptome der PH sind unspezifisch. Die Diagnose wird aus diesem Grund häufig spät gestellt. Ein Leitsymptom ist die Anstrengungsdyspnoe. Weitere Symptome sind Müdigkeit, Gewichtszunahme oder periphere Ödeme, sowie in fortgeschrittenem Stadium Brustschmerzen, Palpitationen, Synkopen, Hämoptysen und Heiserkeit (Dilatation des Truncus pulmonalis). Der Schweregrad wird (analog NYHA bei Herzinsuffizienz) in WHO Funktionsklassen eingeteilt (diese Einteilung wird vor allem bei der PAH verwendet).
WHO Funktionsklassen
| Funktionsklasse I | keine Beschwerden im Alltag, normale Leistungsfähigkeit |
| Funktionsklasse I | Beschwerden ab normaler körperlicher Anstrengung, leicht eingeschränkte Leistungsfähigkeit |
| Funktionsklasse III | Beschwerden bei leichter körperlicher Anstrengung, deutlich eingeschränkte Leistungsfähigkeit |
| Funktionsklasse IV | Beschwerden in Ruhe, Zeichen der Rechtsherzbelastung |
Klinische Zeichen der Pulmonalen Hypertonie
Herzauskultation:
- Akzentuierte pulmonale Komponente des 2. Herztones
- Systolikum über der Trikuspidalklappe (TK-Insuffizienz)
- 3. Herzton
- Diastolikum (Pulmonalklappeninsuffizienz)
Meist erst im fortgeschrittenen Stadium:
- Positiver hepatojugulärer Reflux
- Halsvenenstauung
- Hepatosplenomegalie
- Periphere Ödeme
- Aszites
- Zyanose
Zusätzlich können sich Zeichen der zugrundeliegenden kardialen oder pulmonalen Erkrankung finden.
Weitere initiale Untersuchungen
Röntgen-Untersuchung des Thorax
Häufig zeigen sich erweiterte Pulmonalarterien bzw. dilatierte hiläre Gefässe, es kann jedoch auch ein Normalbefund vorliegen. Das Ausmass der PH korreliert nicht mit dem Ausmass der radiologischen Veränderungen. Das Röntgenbild kann zudem Hinweise auf den Mechanismus der PH ergeben (Lungenparenchymerkrankung, kardiale Erkrankung).
EKG
Nicht diagnostisch, aber evtl. hinweisend.
Zeichen der Rechtsherzbelastung:
- R, ST-Senkungen und T-Negativierung in V1
- Rechtstyp oder überdrehter Rechtstyp
- P pulmonale (P > 0.25 mV in II)
- SIQIII-Typ
- RSB
Falls ein Vorhofflimmern vorliegt, ist die Wahrscheinlichkeit sehr hoch, dass es sich um eine PH bei Linksherzerkrankung handelt.
Echokardiografie
Die Untersuchung wird als Screening-Methode der Wahl eingesetzt und erfasst zudem eine Linksherzpathologie als Ursache der PH (PH Gruppe 2). Die Wahrscheinlichkeit des Vorliegens einer PH wird basierend auf der maximalen Geschwindigkeit der Trikuspidalklappeninsuffizienz (peak tricuspid regurgitant velocity, peak TRV; erlaubt eine grobe Abschätzung des systolischen pulmonalarteriellen Drucks, sPAP) und indirekten Zeichen einer relevanten PH eingeschätzt. Eine genaue Bestimmung des mPAP (zwingend notwendig für die Diagnose einer PH, vgl. Definition) ist mittels Echokardiografie nicht möglich. Die Abschätzung des rechtsatrialen Drucks ist unzuverlässig. Auf eine Addition eines geschätzten rechtsatrialen Drucks zum basierend auf der peak TRV geschätzten sPAP soll daher explizit verzichtet werden.
Wichtig: trotz neuer Definition der PH gemäss Rechtsherzkatheter sind die Grenzwerte für die peak TRV zur Abschätzung der Wahrscheinlichkeit einer PH unverändert geblieben.
Indirekte Zeichen einer relevanten PH:
- Rechtsventrikuläre (RV) Dilatation: RV Durchmesser > linksventrikulärer (LV) Durchmesser im Vierkammerblick an der Basis
- «D-shape» des LV als Ausdruck der RV-Druckbelastung
- Neu (2022): Abnormes «RV-PA Coupling»: Ratio tricuspid annular plane systolic excursion (TAPSE)/sPAP < 0.55 mm/mmHg
- Akzelerationszeit im rechtsventrikulären Ausflusstrakt < 105 ms und/oder midsystolischer Notch im PW-Dopplerprofil
- Rechtsatriale Dilatation: rechtsatriale Fläche im Vierkammerblick > 18 mm2
- Breite und Atemvariabilität der Vena cava inferior: > 21 mm und verminderter inspiratorischer Kollaps
- Dilatation des Truncus pulmonalis: > 25 mm oder grösser als Aorta
Wahrscheinlichkeit für das Vorliegen einer PH aufgrund der Echokardiografie
| PH unwahrscheinlich | Peak TRV ≤ 2.8 m/s ohne indirekte Zeichen einer PH |
| PH möglich | Peak TRV ≤ 2.8 m/s mit indirekten Zeichen einer PH
oder Peak TRV 2.9–3.4 m/s ohne indirekte Zeichen einer PH |
| PH wahrscheinlich | Peak TRV 2.9–3.4 m/s mit indirekten Zeichen einer PH
oder Peak TRV > 3.4 m/s mit/ohne indirekte Zeichen für eine PH |
Falls die Peak TRV nicht messbar (kein Signal oder kein auswertbares Signal) ist, ist eine PH nicht ausgeschlossen. In diesem Fall kommt den indirekten Zeichen entscheidende Bedeutung zu.
Kardiales MRI
Das kardiale MRI hat bei der Diagnostik/Risikostratifizierung der PH zur Bestimmung der Dimensionen der rechtsseitigen Herzhöhlen an Bedeutung gewonnen, was echokardiografisch nur bedingt möglich ist. Verschiedene MRI-Parameter werden neu zur systematischen Risikostratifizierung bei PAH eingesetzt (RVEF, RV end-systolischer Volumen-Index, Stroke Volume Index), vgl. unten.
Rechtsherzkatheter-Untersuchung
Diese Untersuchung ist der Gold-Standard für die definitive Diagnosestellung einer PH. Sie dient auch dazu, den hämodynamischen Mechanismus der pulmonalen Hypertonie zu definieren (prä- oder post.kapillär, vgl. oben) und die PH in eine klinische Gruppe einzuteilen (dazu sind aber oft weitere Untersuchungen notwendig). Eine spezifische Therapie mit pulmonalen Vasodilatatoren darf nur auf der Basis einer Rechtsherzkatheter-Untersuchung durchgeführt werden. Der Rechtsherzkatheter liefert auch wichtige prognostische Parameter, die bei der Risikostratifizierung bei PAH verwendet werden (rechtsatrialer Druck, Cardiac Index, Stroke Volume Index, gemischtvenöse Sättigung).
Ob eine Rechtsherzkatheteruntersuchung durchgeführt werden soll, hängt ab von a) der Wahrscheinlichkeit einer relevanten PH und b) den therapeutischen Konsequenzen bei Vorliegen einer definitiven Diagnose. Bei mittlerer und hoher Wahrscheinlichkeit einer PH und Hinweisen auf Risikofaktoren für eine PAH und CTEPH soll eine Rechtsherzkatheteruntersuchung erfolgen, da sich therapeutische Konsequenzen ergeben (spezifische medikamentöse Therapie bzw. Operation/Intervention/medikamentöse Therapie). Bei mittlerer oder hoher Wahrscheinlichkeit einer PH im Kontext einer Linksherzerkrankung (Gruppe 2) oder Lungenerkrankung (Gruppe 3), gibt es keine Indikation für eine spezifische medikamentöse Therapie. Die Therapie ist immer die Therapie der zugrundeliegenden Erkrankung, wozu kein Rechtsherzkatheter notwendig ist. Entsprechend besteht in dieser Konstellation abgesehen von unklaren Fällen keine Indikation für einen Rechtsherzkatheter. Sinnvoll ist aber ein Follow-up mittels Echokardiografie. Bei unklarer Rechtsherzbelastung muss immer auch an einen Links-Rechts-Shunt gedacht werden. Bei entsprechendem Verdacht TEE/CT (Vorhofseptumdefekt, fehlmündende Lungenvenen) und Rechtsherzkatheter.
Ergänzende Spezialtechniken wie Vasoreaktivitätstestung, Belastung oder Volume Challenge werden in speziellen Situationen durch das Untersuchungsteam ausgewählt.
Lungenfunktionstest (mit CO-Diffusionskapazität), 6-Minuten-Gehtest und Spiroergometrie
Eine grosse Lungenfunktionsprüfung ist integraler Bestandteil der Abklärung bei möglicher PH, zum einen zur genauen Diagnose von obstruktiven und restriktiven Lungenerkrankungen (PH Gruppe 3), zum anderen zum Ausschluss einer relevanten Pneumopathie als Ursache einer PH. Die CO-Diffusionskapazität ist oft vermindert, was aber unspezifisch ist (bei PAH, aber auch Gruppe 3 und teils auch Gruppe 2).
Der 6-Minuten-Gehtest dient zur Bestimmung der Leistungsfähigkeit, der Prognose und der Verlaufskontrolle. Er wird typischerweise bei Patienten mit PAH eingesetzt werden, ist aber auch bei anderen PH-Gruppen etabliert.
Die Spiroergometrie hilft in der Differentialdiagnose von Dyspnoe/Leistungsintoleranz. Bei Verdacht auf eine PH, kann die Spiroergometrie Hinweise auf die PH Gruppe ergeben. Zudem kann so das Ausmass der funktionellen Einschränkung erfasst werden, und es werden wichtige prognostische Parameter erfasst (maximale O2-Aufnahme, ventilatorische Effizienz). Entsprechend wird die Spiroergometrie auch als Verlaufsuntersuchung bzw. Risikostratifizierung im Verlauf eingesetzt.
CT-Thorax mit Kontrastmittel (Lungenembolie-Protokoll oder Dual-Energy) und Ventilations-/Perfusionsszintigrafie der Lunge
Diese Untersuchungen sind zur Diagnosestellung von interstitiellen Lungenerkrankungen, Lungenemphysem, Lungenembolien und anderen Pathologien hilfreich. Zum Nachweis chronischer Lungenembolien ist die Lungenszintigrafie diagnostischer Standard, alternativ kann ein Dual-Energy CT eingesetzt werden.
Basis-Laboruntersuchungen
Bei der Abklärung hinsichtlich der Genese einer PAH gehört eine Basis-Laborabklärung dazu, welche je nach Klinik und Verdachtsdiagnose weiter ergänzt wird.
Basis-Labor:
- Blutbild
- Elektrolyte
- Leber- und Nierenwerte
- Gerinnungsabklärung (primär Suche nach Anti-Phospholipid-Antikörper-Syndrom bei PH Gruppe 4)
- «Rheuma-Profil» (ANA, anti-Centromer, anti-SS-A)
- HIV-Test
- Hepatitis-Serologie
- TSH
- BNP oder NT-proBNP
- Arterielle Blutgasanalyse (aBGA)
- Eisenstatus
Screening von Risikogruppen für eine Pulmonale Hypertonie
Risikogruppen für eine PH sollten regelmässig mittels Echokardiografie gescreent werden
- Bei Sklerodermie und anderen mit PAH-assoziierten rheumatologischen Erkrankungen (SLE, RA)
- Genetisch bedingte PAH (bei direkten Familienangehörigen)
- kongenitale Shunt-Vitien
Diagnostischer Ansatz bei Verdacht auf Pulmonale Hypertonie
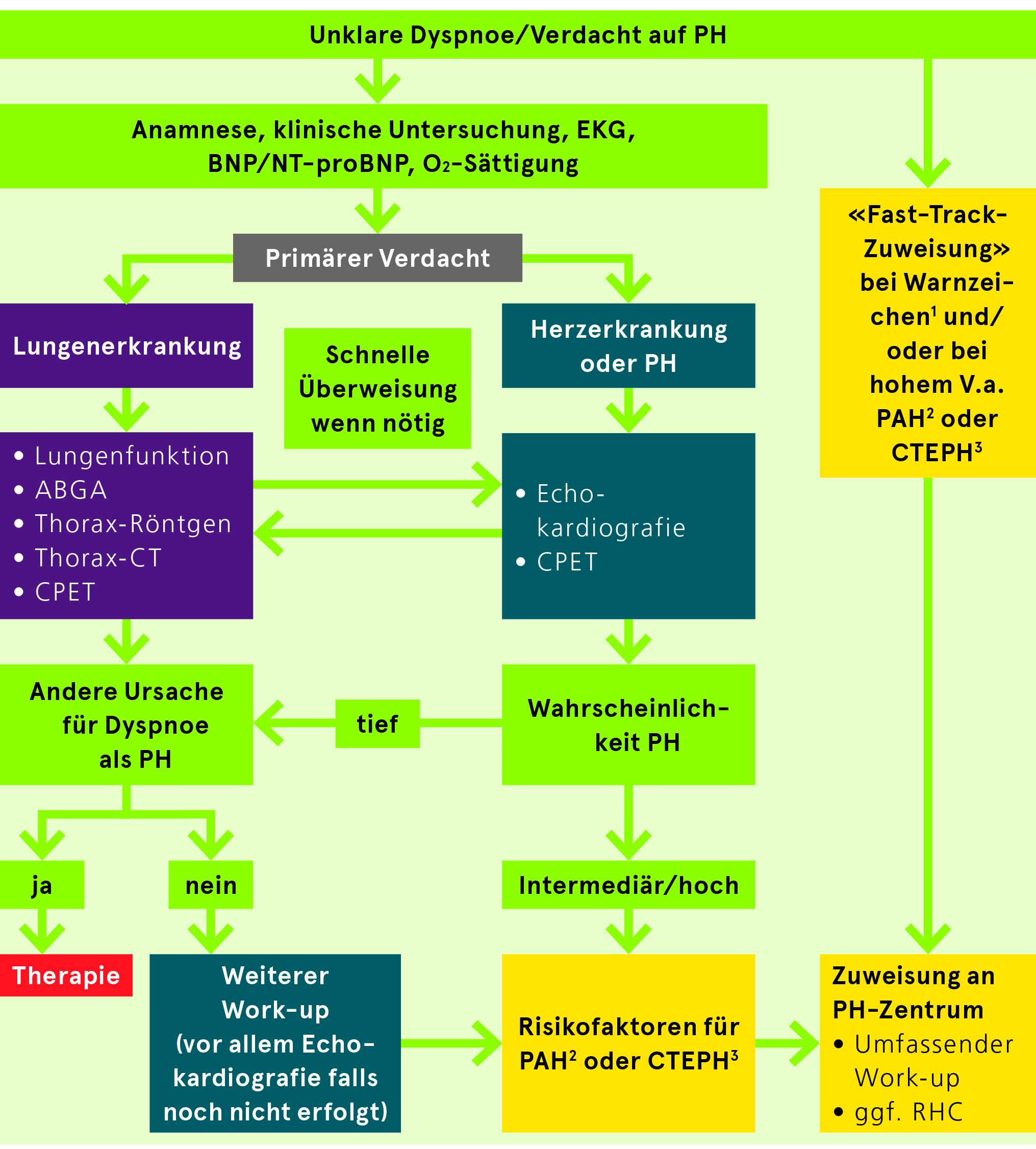
Follow-up und Risikostratifikation (spezifisch für PAH etabliert)
Die Erhebung des Risikostatus ist essentiell (in der Regel alle 3–6 Monate). Die Baseline-Risikostratifikation erfolgt gemäss 3-Strata-Model (tief, intermediär, hoch) und der Follow-Up gemäss 4-Strata-Model (tief, intermediär-tief, intermediär-hoch, hoch).
Folgende Parameter sind im neuen (2022 ESC/ERS Leitlinien) 3-Strata-Model enthalten. Bei Baseline sollen allen Untersuchungen durchgeführt werden (Spiroergometrie fakultativ). Für die Verlaufsbeurteilung im 4-Strata-Model werden alle Messungen ausser Spiroergometrie und Rechtsherzkatheter standardmässig durchgeführt, letztere nur bei klinischer Verschlechterung/unklarer Situation.
- WHO-Funktionsklasse
- Klinischer Verlauf
- Klinische Zeichen der Rechtsherzinsuffizienz
- Anamnese von Synkopen
- BNP oder NT-proBNP
- EKG
- Spiroergometrie
- aBGA oder Pulsoxymetrie
- 6-Minuten-Gehtest
- Echokardiografie
- Kardiales MRI (als Alternative zur Echokardiografie)
- Rechtsherzkatheter
Baseline Risikostratifikation (2022 ESC/ERS Leitlinien)
| Prognostische Faktoren / geschätzte 1-Jahres-Mortalität | Tiefes Risiko < 5% | Intermediäres Risiko 5–20% | Hohes Risiko > 20% |
| Klinische Zeichen der Rechtsherzinsuffizienz | Nicht vorhanden | Nicht vorhanden | Vorhanden |
| Progression der Symptome | Nein | Langsam | Schnell |
| Synkope | Nie | Ja | Rezidivierend |
| WHO funktionelle Klasse | I, II | III | IV |
| 6MWD | > 440 m | 165–440 m | < 165 m |
| Spiroergometrie | Peak VO2 > 15 ml/min/kg (> 65% Soll)
VE/VCO2 slope < 36 |
Peak VO2 11–15 ml/min/kg (35–65% Soll)
VE/VCO2 slope 36–44 |
Peak VO2 < 11 ml/min/kg (< 35% Soll)
VE/VCO2 slope > 44 |
| Natriuretische Peptide | BNP < 50 ng/l
NT-proBNP < 300 ng/l |
BNP 50–800 ng/l
NT-proBNP 300–1100 ng/l |
BNP > 800 ng/l
NT-proBNP > 1100 ng/l |
| Echokardiografie | RA-Fläche < 18 cm2
Kein Perikarderguss TAPSE/sPAP > 0.32 mm/mmHg |
RA-Fläche 18–26 cm2
Kein/minimaler Perikarderguss TAPSE/sPAP 0.19- 0.32 mm/mmHg |
RA-Fläche > 26 cm2
Perikarderguss TAPSE/sPAP < 0.19 mm/mmHg |
| Hämodynamik | RAP < 8 mmHg
CI ≥ 2.5 l/min/m2 SvO2 > 65% SVI > 38 ml/m2 |
RAP 8–14 mmHg
CI 2.0–2.4 l/min/m2 SvO2 60–65% SVI > 31-38 ml/m2 |
RAP > 14 mmHg
CI < 2.0 l/min/m2 SvO2 < 60% SVI < 31 ml/m2 |
| Kardiales MRI | RVEF > 54%
SVI > 40ml/m2 RVESVI < 42 ml/m2 |
RVEF 37-54%
SVI 26-40 ml/m2 RVESVI 42-54 ml/m2 |
RVEF < 37%
SVI < 26 ml/m2 RVESVI > 54 ml/m2 |
Vereinfachtes 4-Strata Risikoassessment für Follow-Ups
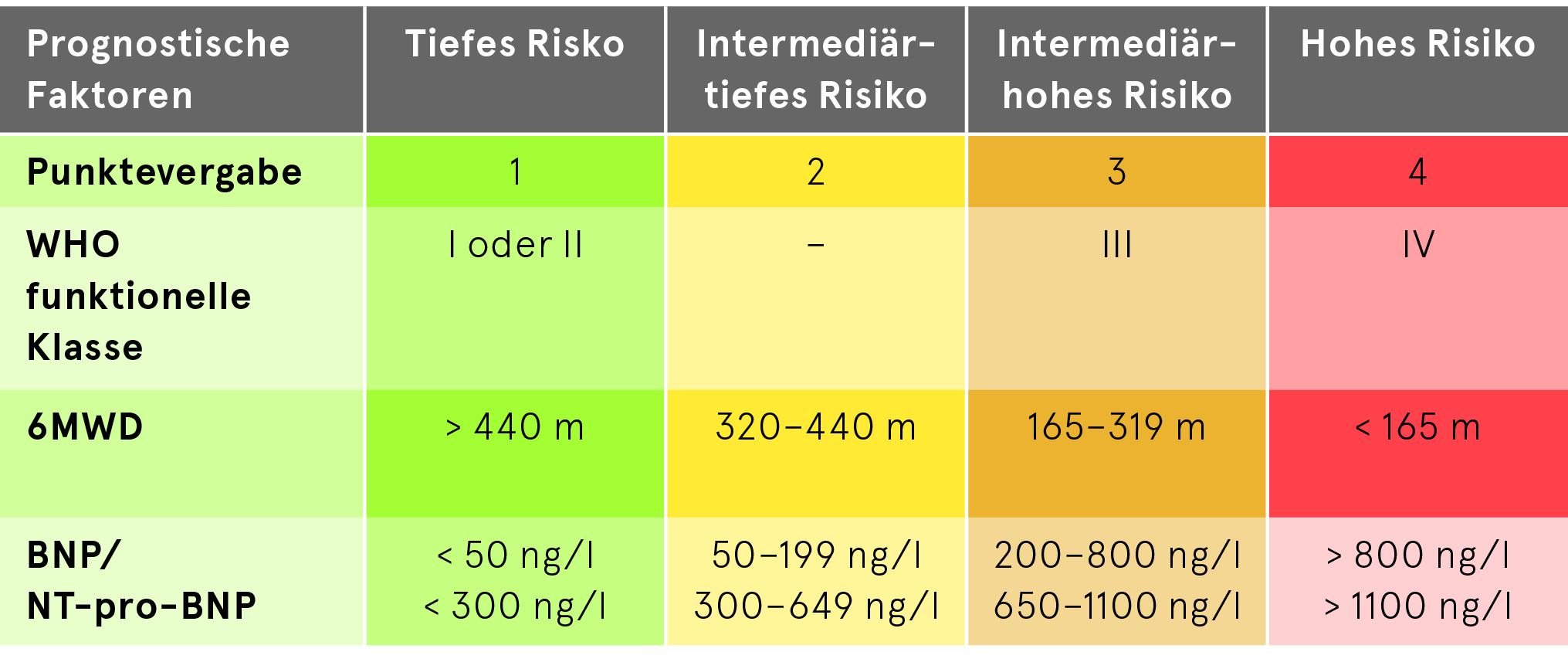
Therapie der PAH
Allgemeine Massnahmen
- Therapie der Grunderkrankung (Shunt-Vitien, HIV, Autoimmunerkrankungen usw.)
- Rauchstopp
- Grippe- und Pneumokokken-Impfung sowie Impfung gegen SARS-CoV-2
- Höhenexposition vermeiden (> 1500 m ohne O2)
- Kontrazeption/keine Schwangerschaft (meist); auf jeden Fall Information der Patientinnen über die Risiken einer Schwangerschaft
- Verzicht auf NSAR
- Psychosoziale Betreuung
- Kardiopulmonale Rehabilitation in spezialisierten Zentren
Medikamentöse Therapie allgemein
- Sauerstoff falls Hypoxämie (aBGA in Ruhe: pO2 < 8 kPa)
- Orale Antikoagulation (bei PH Gruppe 4)
- Diuretika bei Zeichen der Hypervolämie/rechtsventrikulären Dysfunktion
- Behandlung eines Eisenmangels falls assoziiert mit Anämie
Spezifische Therapie (Vasodilatatoren)
Es existieren verschiedene Medikamente aus den drei Substanz-Gruppen (Beeinflussung der Endothelin-, Phosphodiesterase- oder Prostazyklin-Achse) mit nachgewiesenem Effekt auf Leistungsfähigkeit und Prognose. Ziel ist es, gemäss Risikostratifizierung ein «tiefes Risiko» zu erlangen bzw. beizubehalten. Idealerweise sollte die spezifische Therapie früh im Krankheitsverlauf begonnen werden (frühe Diagnosestellung wichtig). Kombinationstherapien (zwei bis drei Substanzklassen) sind heute Standard, werden aber immer individuell festgelegt. Die Indikationsstellung für diese Medikamente erfordert immer einen vollen Work-up inklusive Rechtsherzkatheter. Alle Therapieentscheide werden am interdisziplinären PH-Board besprochen.
Operative Therapieverfahren
Bei der CTEPH (PH Gruppe 4) ist die pulmonale Thrombendarteriektomie (PEA) die Therapie der Wahl. Als Basis für die Operabilität muss eine standardisiert durchgeführte Pulmonalis-Angiografie erfolgen. Es besteht zudem die Option der Ballondilation der Pulmonalarterien für inoperable Patienten. Patienten können hierfür am Swiss-SSPH-CTEPH-Board angemeldet werden (Videokonferenz; Teilnahme aller durchführenden Zentren in der Schweiz).
Bei High-risk-Situation sollte frühzeitig über die Möglichkeit einer Lun- gentransplantation informiert werden, und Patienten sollen dem Lungentransplantationszentrum am Universitätsspital in Zürich zugewiesen werden.
Board für pulmonale Hypertonie am KSSG
Am KSSG findet alle 4 Wochen das PH-Board zur Besprechung von Patienten mit pulmonaler Hypertonie (virtuelles Meeting) statt.
Die Anmeldung für ambulant und stationär betreute Patienten am KSSG erfolgt elektronisch im Boards-Tool (via Medfolio → Module → Boards c37 → Anmeldung) durch den Arzt, welcher den Patienten vorstellt, unter: www.hcweb.ch (Nur mit Login KSSG)
Externe Ärzte sind am Board willkommen. Dafür ist eine Anmeldung des Patienten an das Ambulatorium des Lungenzentrums mit entsprechenden Unterlagen (Echobefunde, Labor, Lungenfunktion, CT-Thorax etc., falls vorhanden) im Voraus notwendig an: lungenzentrum@kssg.ch
Quelle/Link
- Humbert M et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022 Oct 11;43(38):3618-3731., https://doi.org/10.1093/eurheartj/ehv317
Prof Dr. Micha Maeder
PD Dr. Thomas Neumann
Dr. Susanne Pohle
Prof. Dr. Otto Schoch
Dr. Daniel Weilenmann