Chapters
4
Overview
After heart disease, cancer is the leading cause of death in North America. Broadly defined, it is a genetic disease, in which the normal controls governing cellular growth, differentiation, and behaviour are disordered. We can summarize the essential characteristics of cancer cells as follows (Figure 58):

Figure 58: Characteristics of cancer cells
Immortality refers to the fact that, unlike normal cells which replicate a finite number of times, cancer cells undergo mitosis indefinitely. Altered morphology refers to the fact that most cancer cells acquire different structural features and microscopic appearance. Finally, altered behaviour refers to loss of functions such as contact inhibition: when normal cells make contact with each other, they cease proliferating and growing, or enter cell cycle arrest (covered later in this unit). During malignant transformation, uncontrolled cell division occurs, resulting in solid tumour masses.
In this chapter, we will summarize the central paradigm of cell biology, that genes provide the blueprint for the building blocks of cellular activity. This will be followed by a description of the functional and structural differences between normal and cancerous cells, an exploration of the theories of cancer causation, and finally a summary of the systemic signs and symptoms of cancer.
Learning Outcomes
4.1: Articulate the basic structure of a gene, and how genes are translated and transcribed into proteins
4.2: Discuss the mechanisms regulating the cell cycle and cell differentiation
4.3: List the main features of benign and malignant neoplasia
4.4: Summarize some of the main mechanisms underlying carcinogenesis
4.5: Describe the tissue and systemic manifestations of cancer
Key Concepts
• Key features of cancer cells include immortality, altered morphology, and altered cell behaviour.
• Cell growth is regulated by growth factors which activate gene transcription and translation.
• Dysregulated cell growth is the result of genetic alterations in growth-inducing and growth-suppressing genes.
• Oncogenic stimuli include chemical, viral, and radiation-induced changes in DNA.
• Epigenetic modifications to DNA include methylation and histone modifications.
• Morphological changes in cancer cells include altered nuclear size and shape, altered and/or dedifferentiated cell shape, and high mitotic index.
• The most common sign of cancer is cachexia; the most common symptom is fatigue.
Learning Units
Unit 4.2 Dysregulated Cell Growth
Unit 4.3 Features of Cancer Cells and Tissues
Unit 4.4 Systemic Signs of Cancer and Cancer Biomarkers
Unit 4.1: Regulation of Cell Growth

The pathophysiology of cancer is fundamentally an alteration in cellular activity. As we begin this last chapter, it is helpful to briefly review how cellular growth and activity is regulated, starting with the central dogma of cell biology: that DNA (deoxyribonucleic acid) carries the genetic code or blueprint for proteins, which in turn help to regulate cellular activity.
The basic unit of DNA is a nucleotide, which is a molecule consisting of a 5-carbon sugar (ribose) with an attached phosphate molecule, and one of four different nucleobases: adenine, cytosine, guanine, and thymine (Figure 59).
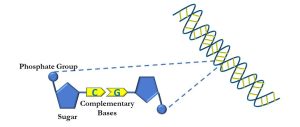
Figure 59: Primary structure of DNA
Nitrogenous bases form complementary pairs via hydrogen bonding (cytosine binds to guanine, adenine to thymine). Adjacent nucleotides form polynucleotide sequences which ultimately comprise the alpha double helix that is DNA. While DNA is understood to form the basis for chromosomes, it is important to remember that DNA is usually tightly packed by associating with histone proteins. When DNA is either replicated or the genetic code is being read, it is unwound and ‘unzippered’, as depicted below, into sense and antisense strands (Figure 60).
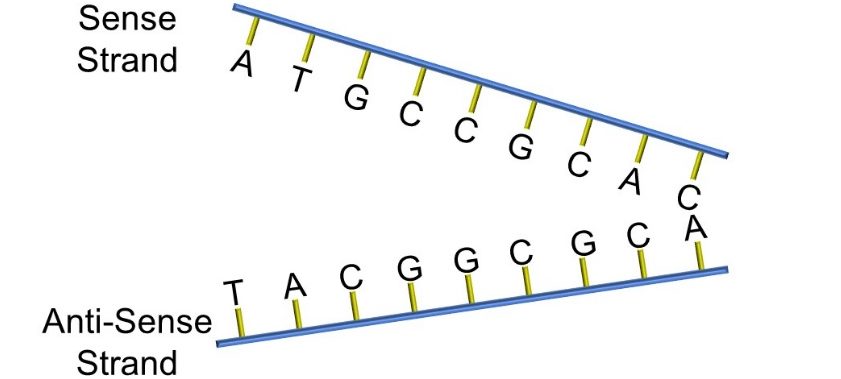
Figure 60: Sense and anti-sense strands of DNA
The actual genetic code is carried by the sequence of nucleotides in DNA. Each sequence of nucleotides (eg: ATG) corresponds to a complementary sequence in messenger ribonucleic acid (mRNA). For example, ATG corresponds to ‘UAC’ on mRNA, and ultimately this codes for the amino acid tyrosine, while GGC codes for glycine and GUG for valine (Figure 61).

Figure 61: The genetic code
Note that the nucleobase thymine (T) is replaced in mRNA with another nucleobase, uracil (U).
The molecular machinery of the cell utilizes this information to produce small amino acid sequences called peptides and longer sequences called proteins. This is a multi-step process, beginning with the production of single-stranded mRNA within the nucleus, a process referred to as transcription. After mRNA leaves the nucleus, the genetic message is ‘read’ or translated by ribosomes, during which individual amino acids are carried by transfer RNA (tRNA) (Figure 62).

Figure 62: tRNA and mRNA
The triplet of UAC on the mRNA molecule is referred to as a codon. The corresponding nucleobases on the specific, paired tRNA molecule are referred to as an anticodon, and a tRNA with this anticodon carries a specific amino acid, in this case tyrosine.
The pairing of mRNA codons with tRNA anticodons occurs in the cytoplasm at specific sites on ribosomes. As the codon on mRNA matches with its corresponding tRNA, the mRNA moves through the ribosome, making available the next codon of triplet bases. During this process, called translation, amino acids become covalently linked into a growing polypeptide chain (Figure 63).

Figure 63: Transcription and translation
Peptides and proteins function as either structural components of the cell or enzymes. These molecules in turn catalyze chemical reactions and regulate cellular growth and differentiation. Both transcription and translation must be tightly regulated for normal cellular functioning. Transcription, for example, is regulated by proteins called transcription factors, which bind to specific nucleotide sequences at the start of the coding sequence of a gene called the promoter. Transcription factors enable the gene to be read by another protein, called RNA polymerase. At the end of the gene is another sequence of nucleobases which constitute a ‘stop’ sequence called the terminator (Figure 64).
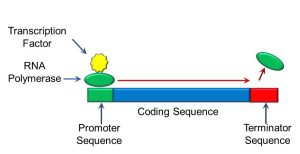
Figure 64: Gene transcription
The Cell Cycle
At a fundamental level, cancer is a pathology affecting cell differentiation and growth. Given this fact, a good place for us to start examining this complex disease is to understand the processes which control cell growth and differentiation. Somatic (‘of the body’) cells replicate and divide by the process of mitosis, in which genetic material is replicated and one parent cell divides into two ‘daughter’ cells. In contrast, when germ cells divide, genetic material is halved, a process known as meiosis. Beyond this basic summary, we will not concern ourselves further with the mechanisms and details of mitosis and meiosis. Instead, we will focus on the period between mitosis, called the cell cycle, as depicted below (Figure 65):

Figure 65: The cell cycle
The cell cycle is divided into four phases: G1, S, G2 and M. ‘G’ stands for ‘gap’, ‘S’ represents ‘synthesis’, and ‘M’ stands for ‘mitosis’. During mitosis, two processes occur: karyokinesis and cytokinesis. Karyokinesis is the process during which double-stranded chromosomes separate into separate nuclear envelopes, while cytokinesis refers to the process of cell division. After mitosis is complete, two identical daughter cells are produced, which then enter the G1 phase. During G1, cells grow and differentiate into their final forms, and, later in G1, duplicate much of the cellular contents, with the exception of chromosomes.
In the event that conditions do not favor cell growth, cells become arrested in G0, where no further growth or differentiation occurs. Some cells, such as muscle and nerve, do not usually divide after differentiation. Others, such as epithelial cells and some connective tissue cells, are capable of cell divisions. The latter progress further into the cell cycle after arriving at the ‘R’ or ‘restriction point‘, which can be considered the ‘go/no go’ signal for eventually entering mitosis, which is preceded by the S phase. During S phase, single-stranded chromosomes duplicate themselves, becoming double-stranded. It is important to note that this does not involve an increase in the number of chromosomes. After S phase, cells enter the second gap or G2 phase, during which the cell prepares for mitosis by synthesizing the materials needed for cell division, and checks for errors in the duplicated chromosomes. Once complete, the cell enters mitosis.
Growth Factors
Progression through the cell cycle, including replication and differentiation, involves a variety of growth-promoting factors; examples of these include epithelial growth factor (EGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF). Most of these factors bind to extracellular receptors, and activate molecular intermediates including G proteins (eg: ras) and kinases which transduce, or transmit, the growth factor signal. The latter typically phosphorylate inhibitory complexes which activate nuclear transcription factors (eg: myc, fos, jun). As described previously, transcription factors bind to genes which have specific promoter sequences, which begin the processes of transcription. Ultimately, the ‘building blocks’ of life, such as structural proteins and enzymes, are produced as part of the process of growth and differentiation. The sequence of growth receptor activation, transduction, and nuclear transcription are summarized in Figure 66:
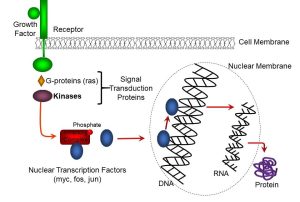
Figure 66: Growth factors
We have thus far discussed how cell growth is normally stimulated. In the next section of this chapter, we will discuss mechanisms underlying dysregulated cell growth, which is one component of neoplasia.
Unit 4.2: Dysregulated Cell Growth

As you might expect, the progression of cells through the cell cycle is under a series of tight regulatory controls. As we begin to discuss the biology of cancer, it will become evident that dysregulation or disruption of these controls can lead to alterations in cell growth and differentiation. We can summarize our discussion of regulators of the cell cycle with the notion that it is under the control of a system of “Stop” and “Go” signals(Figure 67).

Figure 67: Dysregulated cell growth and neoplasia
Two possible scenarios may lead to dysregulated cell growth and, ultimately, carcinogenesis: either a pathway that normally promotes cell growth may become disinhibited, or ‘turned on’, or a process which stops cell growth may become permanently disactivated. In the former scenario, a receptor such as EGF is altered, and activates in the absence of its ligand. Other parts of the pathway, such as receptors, transducers (Chapter 1.2), or transcription factors (Chapter 4.2), may be activated or over-expressed (too much of the protein is made). Conversely, a signaling pathway that stops cell growth may become knocked out (silenced).
In general, two types of genes are implicated in cancer: proto-oncogenes and tumour suppressor genes.
Proto-oncogenes
Proto-oncogenes are genes that confer an advantage to tumours though gain-of-function mutation. By extension, the activated counterparts of proto-oncogenes are oncogenes. Some examples include:
- HER2/neu: a growth factor receptor; breast cancer
- B-raf: a signal transduction gene associated with malignant melanoma
- Myb: a transcription factor that is activated during leukemia
Tumour Suppressor Genes
Tumour suppressor genes confer an advantage to a tumour though loss-of-function mutation. These are actually more common than oncogenes, and examples include the following:
- Rb (retinoblastoma): this is a gene we have mentioned earlier, and its protein product is a cell cycle regulator; it is a causal factor in neuroblastoma
- APC (adenomatous polyposis): the protein product is a cell adhesion protein, and its expression is reduced in some colon cancers
- BRCA1&2 (BReast CAncer Gene): this is a DNA repair gene that is mutated in many breast cancers
These mechanisms help us to explain why cellular replication and growth become dysregulated. However, one of the other main characteristics of cancer cells that we’ve mentioned at the beginning of this unit is immortality. Normal cells do not typically divide indefinitely; at some point, cells stop dividing. One theory is that cells ‘wear out’ and become unable to divide.
Cellular Immortality
One of the mechanisms behind this involves an enzyme called telomerase. To understand what this enzyme does, we need to first describe the basic structure of a chromosome, which contains tightly-packed DNA, as well as regions which do not contain any genetic information, including centromeres and telomeres (Figure 68).

Figure 68: Telomeres and centromeres
The center of a chromosome is called the centromere, which serves as an attachment point for cytoskeletal proteins during mitosis. At either end of a chromosome are multiple telomeres (‘telos’ being Greek for ‘distant’). Telomeres are nucleoprotein repeats which contain no coding information.
Normally, during cell division, telomeres are lost, likely due to mechanical wear (Figure 69).

Figure 69: Telomere loss during normal cell division
After enough cell divisions, coding regions of the chromosome are also lost, and this is likely one reason why non-cancerous cells can only divide a finite number of times. In certain types of ovarian cancer, an enzyme known as telomerase is over-expressed. Telomerase enzyme replaces telomeres on the ends of chromosomes, and is normally expressed at low levels. We now know that telomerase is over-expressed in some cancers, such as some ovarian cancers.
Oncogenesis
As alluded to above, cancer is a genetic disease—that is, cancer is usually the result of changes to genes that control cellular growth and differentiation, function and/or genetic repair mechanisms. Alterations in DNA leading to cancer is referred to as oncogenesis, and may be the result of several factors (Figure 70):

Figure 70: Mechanisms of oncogenesis
Viral transformation occurs when viral DNA is stably inserted into the genome (the repetoire of genes in an organism), affecting the growth and/or phenotype (physical and/or behavioural characteristics) of cells. Some viruses are particularly effective at transforming cells, and can be considered to be oncogenic. These viruses include: hepatitis B and C, human papilloma virus (HPV), Epstein-Barr virus (EBV), human herpes virus 8 (HHV8), Merkel cell polyoma virus (MCPV), and HTLV-1. Approximately 20% of cancers are believed to be caused by these viruses. The mechanisms by which these viruses cause cancer are complex, and may involve causing chronic inflammation, interfering with DNA repair mechanisms, alteration of ribosomes, and cell cycle alterations.
Genomic mutation may be caused by viruses, but may also be caused by chemical or other agents called mutagens. Of the hundreds of thousands of chemicals that humans are exposed to, less than 5% have been investigated for their mutagenic potential. Despite this lack of information, many chemicals have been identified as mutagenic and/or carcinogenic, including polyaromatic hydrocarbons (PAHs). PAHs include compounds such as benzo-[a]- pyrenes. These chemicals may alter DNA by either of two mechanisms: (a) generating free radicals; and (b) forming metabolic by products called DNA adducts, which physically stick to DNA (Figure 71).
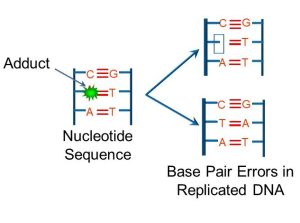
Figure 71: DNA adducts
If free radicals or adducts bind to regions which code for cell cycle regulatory proteins or to DNA repair genes, then these mutations may be expressed as cancers. Alternately, PAHs can activate cellular receptors (such as the estrogen receptor), which stimulates cell division. Other mutagenic (and possibly carcinogenic) chemicals include aniline dyes, which are implicated in bladder cancer, and natural compounds such as aflatoxins which have been linked to liver cancer. One of the conundrums of traditional chemotherapy is that some of the drugs in common use, such as cisplatin and cytochalasin, are themselves mutagenic, and possible carcinogens.
As mentioned earlier, radiation can alter DNA. Ionizing radiation, which includes any radiation that is strong enough to strip electrons from atoms, is particularly effective at causing DNA damage and/or mutation. Examples of ionizing radiation include ultraviolet light, X-rays, gamma rays, and alpha and beta particles. For example, ultraviolet light can induces pyrimidine dimers which have been cited as promoters of skin cancers. Other forms of ionizing radiation are generally known to cause mutations in DNA, and to promote malignancies such as leukemia, thyroid, lung, colon, and breast cancers.
Self-Study Question:
Aside from genetic mutations, in which the nucleotide sequence is altered or mispaired, or oncogenes introduced by viruses, genomic ‘tags’ or modifications made to DNA can also affect the activity of genes. The latter is referred to as epigenetics. Epigenetic modifications include the addition of methyl or acetyl groups to regions of DNA, or changes to proteins that package DNA. Whatever the mechanism, these changes alter the patterns of DNA expression and, in turn, the structure and behaviors of cells and tissues. Irrespective of the initial insult, if a gene is altered that regulates the cell cycle, cell differentiation, or DNA repair, the result may lead to carcinogenesis.
Hereditary Causes of Cancer
Defective chromosomes may be passed from one generation to the next through germ cells, which include sperm and eggs. Stable mutations that occur in germ cells are linked to heritable cancers. For example, retinoblastoma (a cancer of the eye) is due to a anti-oncogene on chromosome 13 that controls cell growth. Examples of other heritable mutations include the following:
- Hereditary breast and ovarian cancer syndrome (HBOC)
- BRCA1&2
- Associated with breast cancers, but also with ovarian pancreatic and prostate cancer
- Lynch syndrome (hereditary nonpolyposis colorectal cancer)
- Mutated mismatch repair (MMR) genes, which repair damaged DNA
- Li-Fraumeni syndrome
- Mutated TP53, a tumour suppressor gene, that has been associated with osteosarcoma, leukemia, brain, adrenal and breast cancers
Carcinogenesis and Tumor Growth
We have thus far discussed how oncogenic stimuli may alter the genome. However, the formation of cancer cells is believed to involve a multi-step process (Figure 72):

Figure 72: Multi-step model of tumorigenesis
Initiation occurs when a normal cell’s proto-oncogenes are activated. Proto-oncogenes regulate early rapid cellular growth of embryonic life and become repressed after birth. An event or exposure that can damage DNA may damage the genes of a normal cell, turning on genes that should remain repressed. These stimuli may be viral, chemical, or physical. After a cell becomes initiated, it usually takes some time before a cell becomes cancerous. The latency period is the time from when a cancer is initiated until it becomes a cancer cell; a substance that shortens the latency period by promoting or enhancing cell growth (eg: hormones, drugs, and many industrial chemicals) is called a promoter. NOTE: this is not the same as the promoter on a gene. Progression is a period of tumour growth until enough of a tumour mass accumulates to form a detectable tumour. Most tumours are not physically detectable until approximately 1 billion cells are present (approximately 1 cm). Until that point, the tumour gets oxygen and nutrients via diffusion through surrounding cells. After a tumour mass reaches 1 cm, it must get its own blood supply through angiogenesis (new blood vessel formation). Angiogenesis may be considered part of progression.
Tumour cells at the centre of a mass are also further from oxygen and metabolites normally supplied by capillary networks. To maintain homeostasis within the tumor, and conditions favorable for growth, new capillaries must be generated. One theory posits that hypoxia, or low oxygen concentration within the tissue mass, may trigger the release of pro-angiogenic factors. Some of these include:
- Epidermal growth factor (EGF)
- Fibroblast growth factor (FGF)
- Platelet-derived growth factor (PDGF)
- Vascular endothelial growth factor (VEGF)
These cytokines have a growth, or trophic effect on endothelial cells (epithelial cells lining blood vessels) and pericytes (cells regulating blood flow) (Figure 73).
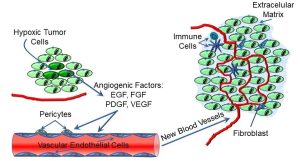
Figure 73: Angiogenesis and tumor growth
Angiogenesis is a complex, multi-step process, and may differ in different tissues, but cytokines like VEGF act on vascular endothelial cells, among others, to promote the growth of new capillaries that supply the tumour. As depicted below, the growing tumour mass often comprises tumour cells, blood vessels, fibroblasts, and immune cells, all within an extracellular matrix. This complex ‘ecosystem’ of tumour, non-tumour, and immune cells and connective tissue becomes capable of maintaining its own homeostatic conditions, often at the expense of ‘normal’ neighbouring cells and tissues. Some research suggests that angiogenesis may also be a precursor to metastasis: VEGF also induces the secretion of proteases, which may help to digest limiting barriers.
Finally, metastasis is the condition in which cancer cells move from the primary site to more remote sites; such growths are called metastatic or secondary tumours. Some metastases are more common than others:
- Primary lung tumours often metastasize to the brain.
- Primary breast tumours frequently form secondary tumours in bone, brain, or liver.
Not all cancer cells are able to metastasize. As depicted below, not all tumour cells survive immune surveillance (depicted by the ‘X’), and only some of the surviving cells acquire through mutation specific traits which allow them to move to secondary sites (Figure 74).

Figure 74: Metastasis
Metastatic cells often have to have the molecular machinery to move, penetrate encapsulating barriers, and/or attach outside the original tumour sites. This can complicate cancer therapy: secondary tumours are often different from the tumour cells at the primary site, where they were initially detected. As well, cancer cells often mutate over time. One example are breast carcinomas, which may shift from being estrogen receptor positive (ER+) to estrogen receptor negative (ER-).
Unit 4.3: Features of Cancer Cells and Tissues
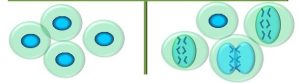
Although there are many forms of cancer, we can generalize about the physical and functional traits of cancerous cells. Physically, cancer cells differ from normal cells in the following ways (Figure 75):
- They have larger, irregularly-shaped nuclei
- Cell shape is altered or irregular
- Cancer cells typically are dedifferentiated and/or express unusual markers; and
- They have a high mitotic index

Figure 75: A comparison of normal cells vs. cancer cells
One explanation for the larger nuclei is that, in some cases, cancer cells cycle rapidly through G1 of the cell cycle, and thus do not have enough time to develop. Mutated structural genes may produce proteins that cause alterations in cell structure. Finally, many malignant tumours, especially ones that are considered ‘high grade’, have a high proportion of cells undergoing mitosis (high mitotic index). This is consistent with a loss of cell cycle controls. Tissues which are typically lost and replaced at a high rate, such as epithelial cells, have a high mitotic index compared to non-dividing cells, such as muscle or nerve. This is one of the contributing factors as to why we see more cancers in rapidly-dividing cells: there is a greater likelihood of an error with each progression through the cell cycle. Another reason for higher cancer rates among epithelial cells is that these are more directly exposed to mutagenic environmental stimuli. However, cancer cells in any tissue have an inappropriately higher mitotic index.
In this context, immortality refers to the fact that, unlike normal cells which divide a finite number of times, cancer cells keep dividing more-or-less indefinitely. Cancer cells also usually (though not always) progress rapidly through the cell cycle. We will explore one of the mechanisms underlying these functional changes a little later. Behavioural alteration refers to altered tumour cell activity in comparison to normal cells. For example, many non-cancerous cells exhibit contact inhibition, in which they stop moving or dividing when they come into contact with other cells. This behaviour is often lost in cancerous cells.
At this point, we should pause to discuss what is a ‘tumour’. Not all tumours are cancerous, and in this context we will describe benign and malignant tumours.
Benign tumours
Examples of benign tumours include moles, nerve ganglia, cysts, uterine fibroid tumours, and endometriosis. The characteristics of benign tumours include:
- inappropriate cell growth
- benign tissues unnecessary for normal functions
- strongly resemble parent cells; retain morphology of tissue of origin
- specific differentiated functions
- joined tightly together
- do not migrate
- small nucleus to cytoplasm ratio
Malignant Tumours
In contrast, malignant tumours are characterized by:
- appearance changed from cells of origin
- size and shape make identification of the origin more difficult
- poorly controlled growth continuous cell division and growth
- growth compares to the parent cell
- eg: bone marrow cells divide rapidly, leukemia cells divide rapidly
- large nucleus to cytoplasm ratio
- loss of differentiated functions/appearance
- migrate through blood vessels and tissues
- altered surface proteins
- break away from tumour with only slight pressure
Cancer Staging
Cancer staging is based on some of the characteristics we’ve just discussed. It is one means of standardizing diagnosis, prognosis, and treatment, and predicting tumour aggressiveness and how a cancer may respond to treatment. The following grading system is universally used:
- Grade 1: low grade; cells similar to parent cells
- Grade 2: low to moderate malignancy; cells have some normal some malignant features
- Grade 3: moderate to high-grade malignancy; cells have more malignant features
- Grade 4: high-grade malignancy; cells have no normal cell appearance
Another means of staging tumours is the TMN system:
- T= tumour size
- N= lymph node involvement
- M= metastasis
Letters after headings indicate extent of disease. For example, tumour size may be evaluated as: TX: cannot be evaluated; T0: no evidence of primary tumour; Tis: carcinoma in situ (CIS: abnormal cells are present but have not spread to neighboring tissue; although not cancer, CIS may become cancer and is sometimes called preinvasive cancer). Nodes may be assessed as: NX: regional lymph nodes cannot be evaluated; N0: no regional lymph node involvement; N1, N2, N3: Degree of regional lymph node involvement (# and location of lymph nodes). Finally, metastases are graded as: MX: distant metastasis cannot be evaluated; M0: no distant metastasis; M1: distant metastasis is present
Other, more tumour specific grading schemes also exist. For example, Dukes D is applied to colon cancer invasion of the wall of the bowel, and Clarke’s is a system for grading levels of skin cancer. Brain and spinal cord cancers are staged according to cell type and grade, whereas lymphomas typically are graded according to the Ann Arbor staging classification, and gynecological cancers are graded using the FIGO classification based on TMN. Finally, specific nuclear grading (how much do nuclei look like normal nuclei), hormone receptor status (eg: estrogen/progesterone and Her2/neu status for breast cancers) are other examples of tumour-specific grading.
Microscopic Appearance of Tumours
The microscopic appearance of tumours can vary greatly, and it is the realm of the pathologist to interpret these. However, in the following images we can visualize some of the changes in appearance and organization of benign and malignant tumours (all images are from www.pathologyatlas.ro, an open-source pathology website).
We earlier in this unit described some of the differences between benign and malignant tumours. In the microscopic images below, benign fibroadenomas of the breast are presented (Figure 76).

Figure 76: Benign epithelial tumour of the breast. One can see the following features: Panel A. Stromal (non-secretory) cell proliferation compressing milk ducts. This type of tumour is called an intracanalicular fibroadenoma. Panel B. Fibrous stroma proliferation around milk ducts, in what is classified a pericanalicular fibroadenoma.
In comparison, in the next image we see a higher magnification view of a malignant epithelial breast tumour (Figure 77):
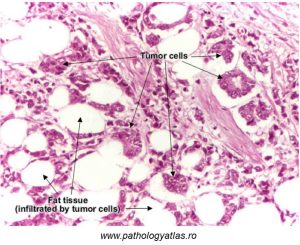
Figure 77: Features of malignant breast epithelial tumors
As with approximately 90% of breast cancers, epithelial cells lining milk ducts have been transformed into cancer cells. These cells appear atypical in morphology, and form ribbons or nests. As with invasive cancer cells, the basement membrane is disrupted. Tumours arising from epithelial cells are termed ‘carcinomas‘.
In the microscope image below (Figure 78), some of the features of a benign epithelial colon polyp are distinguishable.
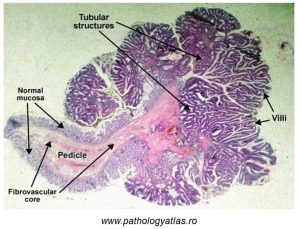
Figure 78: Benign epithelial tumour (colon polyp)
An abnormal epithelium is visible, as manifested by villi which are markedly different. At a higher magnification, cellular dysplasia would be evident (recall Unit 2).
In the next image (Figure 79), a malignant adenocarcinoma of the colon is shown:
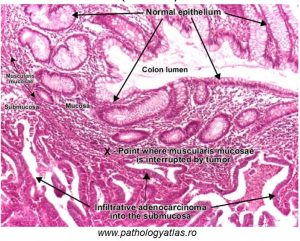
Figure 79: Features of a malignant epithelial tumor of the colon
Adenocarcinomas are epithelial cell cancers which arise in glandular tissue, and in this image of the colorectal mucosa, you can note irregular glands comprised of cells which do not resemble normal, adjacent glandular cells.
In the previous images, we have focused on epithelial tumours, which are more common. However, connective tissue tumours can and do appear frequently, as typified by the image below of an osteosarcoma (Figure 80).

Figure 80: Features of an osteosarcoma
‘Osteo’ refers to bone, and ‘sarcoma’ refers to connective tissue. In this image, tumour cells are pleiomorphic (variable in appearance) and anaplastic (having lost similarity in appearance to the parent cell). Also visible are irregular bony trabeculae.
Finally, cancers of lymphoid cells are very common, and also result in altered cellular morphology. In the microscope image below, cells from the bone marrow of a person with Hodgkin lymphoma is shown. In Hodgkin lymphoma, characteristic Reed-Sternberg cells are visible; these have mirror-image nuclei said to resemble ‘owl eyes’ (Figure 81).
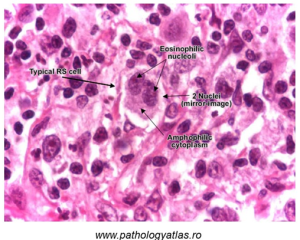
Figure 81: ‘Reed-Sternberg’ (RS) cells characteristic of Hodgkin lymphoma
Self-Study Question:
Unit 4.4: Systemic Signs of Cancer and Cancer Biomarkers
Signs and Symptoms of Cancer
The signs and symptoms of cancer can vary considerably, depending on the type of tumour, infiltration and disruption of specific tissues and organs, and cancer stage. For example, cough, dyspnea, and hemoptysis are commonly associated with lung cancers; occult bleeding and digestive issues are often seen in colorectal cancers; and cognitive/sensorimotor defect are common in brain cancers. However, irrespective of the type of cancer, the most common symptom is fatigue, and the most common sign is cachexia.
Fatigue and Cachexia
In most instances, fatigue is multifactorial: either a negative energy balance secondary to inadequate nutrition, chemotherapy or cancer toxins, and/or poor oxygenation. Cachexia, which comes from the Greek ‘kakos’, meaning ‘bad things,’ and ‘hexus’, meaning ‘state of being’, is manifested by marked loss of body weight, anorexia, and anemia. The extent of cachexia is variably expressed, and pathophysiology of cachexia is also multifactorial. Weight loss may in part be due to reduced nutrient intake; however, the weight loss of cachexia differs from that of starvation in that the latter primarily involves the loss of adipose tissue, whereas cachexia involves equal loss of adipose and muscle. What accounts for this difference? Again, numerous factors are involved, but inflammatory cytokines, such as IL-1, IL-6 and TNF alpha are typically at higher levels in cancer patients, and these can increase muscle proteolysis and affect the activity of hypothalamic neurons which regulate energy expenditure.
Self-Study Question:
Inflammation and Cancer
This presents a convenient opportunity for us to discuss the link between cancer and inflammation. In the 19th century, the physician and scientist Rudolph Virchow recognized the link between inflammation and cancer, noting that cancers often develop at sites of inflammation. A debate has centred on whether inflammation is a cause or simply a manifestation of cancer. The likelihood is high that inflammation is both. As you may recall from earlier in this book, the inflammatory response yields cytokines and generates free radicals. The latter may damage DNA, while cytokines can alter gene expression. Recall that TNF-alpha activates transcription factors, including nuclear factor kappa B (NFκB). In fact, many other stimuli can activate tNFκB, including free radicals. NFκB activates multiple genes, which in turn can increase tumour promotion and progression. A more detailed discussion goes beyond the scope of this chapter, but it is important to note that inflammation is both a marker and a cause of cancer.
Cancer Biomarkers
Plasma and other body fluid markers are also manifestations of cancer. For example, the following isoenzymes have been detected in cancer patients:
- Acid phosphatase (breast, prostate cancer)
- Alkaline phosphatase
- Placental alkaline phosphatase (choriocarcinoma, ovary, breast pancreas)
- Nonplacental alkaline phosphatase (osteogenic sarcoma, parathyroid(
- Galactosyl transferase (lung, breast, esophagus, stomach cancers)
- Aminopeptidases (pancreatic cancer)
- Gamma-glutamyl transpeptidase (cancers of the pancreas, stomach lining, liver)
- Ribonuclease, sialytransferase (pancreatic cancer)
It is important to note that these are relatively non-specific, and not exclusive, manifestations of cancer. In other cases, specific cancer antigens have been associated with cancers:
- Carcinoembryonic antigen (many solid tumours)
- Alpha fetoprotein (liver, ovary, testicular tumors)
- CA 19-9, CA 27-29, CA 15-3 (staging of breast tumours)
- Prostate-specific antigen (prostate cancer)
- Human chorionic gonadotropin (ovarian cancer)
Additionally, ectopic hormones are manifestations of some cancer:
- Adrenocorticotropic hormone (ACTH), antidiuretic hormone (ADH), melanocyte stimulating hormone (MSH) may all be expressed in specific lung tumors
- Thyroid stimulating hormone (choriocarcinoma)
- Insulin (lung tumours)
Again, with the exception of PSA, most of these are relatively non-specific manifestations
Self-Study Question:
Advanced Biomarkers
As biomedical research continues, increasingly specific and sensitive biomarkers of cancer are being developed for particular cancers. We will conclude this unit with a brief look at three molecular markers which have been associated with many cancers.
DNA Methylation
DNA methylation is both a possible causal factor in some cancers, as well as a diagnostic marker. As discussed earlier, methylation can silence a gene, and typically occurs in occurs in the dinucleotide 5′-CpG-3′ (Figure 82).
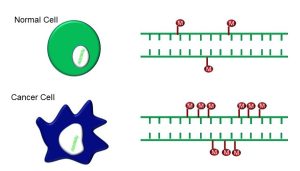
Figure 82: DNA methylation
Many cancers have methylated genes (Table 1):
| Cancer | Gene | Function |
| Breast | TMS1 | Angiogenesis |
| DAPK | Apoptosis | |
| Colon | p14ARF | Cell Cycle |
| hMLH1 | DNA Repair | |
| Kidney | VHL | Transcription |
| Ovarian | BRCA1 | Transcription |
| Prostate | GSTP1 | Detoxification |
| p14ARF | Cell Cycle | |
| Stomach | p14ARF, hMLH1 | Cell Cycle/DNA Repair |
Table 1: Cancer types associated with methylated genes
As you will note, hypermethylation has been detected in genes which regulate most of the processes we have discussed. Hypermethylation affects many of the genes involved in regulating the cell cycle, mitosis, DNA repair, angiogenesis, and metastasis.
MicroRNAs
These are non-coding RNA molecules (they are not translated into proteins), which nonetheless control a myriad of functions, including silencing RNA and regulating gene expression. As a biomarker, microRNAs have many benefits as cancer biomarkers as they are detectable in plasma, serum, urine, and saliva. Moreover, breast, lung, and colorectal cancers all have specific microRNAs which either increase or decrease. As a result, much effort is currently underway to increase the utility of microRNAs as biomarkers for diagnosing specific cancers, or identifying patients more likely to have a particular outcome (prognostic biomarker) or respond to a particular treatment (predictive biomarker) (Figure 83).
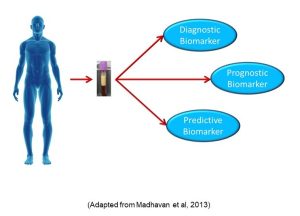
Figure 83: MicroRNA biomarkers
Single Nucleotide Polymorphisms
SNPs are variations in a DNA sequence whereby one nucleotide is different from the rest of the population (Figure 84).

Figure 84: Single nucleotide polymorphisms
A large number of genes associated with certain cancer types have SNPs. In fact, SNPs are the most common type of change in DNA. SNPs can be located in promoter regions as well as in the coding regions of genes, and can affect gene expression. SNPs can not only determine cancer susceptibility, but also may determine how well a cancer will respond to treatment, as well as having great diagnostic potential.
Self-Study Question:
Chapter Summary
a molecule consisting of a nucleoside (a 5-carbon sugar) with a phosphate group
nitrogen-containing compounds which can pair with other compatible nucleobases by means of hydrogen bonding
coding strand of DNA
non-coding strand of DNA
a form of RNA from which genetic information is translated into protein
an organic compound containing a carboxyl group ((-COOH) and an amino group (-NH2) attached to a central carbon atom. The identity of an amino acid is based on what other molecules are attached to the central carbon atom.
a set of three nucleotides in mRNA which correspond to the anticodon on tRNA
a triplet of nucleobases on tRNA; corresponds to specific codons on mRNA
the process by which a sequence of mRNA is converted into a protein
a polymer consisting of a few or many amino acid molecules; forms part (or the whole) of a protein
proteins which control the rate of transcription of DNA into mRNA; specific transcription factors bind to specific DNA sequences
DNA sequences that determine where transcription of a gene by RNA polymerase begins
nucleic acid sequence at the end of a gene; triggers the release of a newly-synthesized mRNA sequence
cells which give rise to gametes (sperm or eggs)
