Chapters
2
Overview
In this unit, we discuss how cells and tissues adapt to the environment. We will also explore the mechanisms and effects of cellular injury, and different mechanisms of cell death.
Learning Outcomes
2.1: Assess cellular adaptations to stress
2.2: Analyze the effectors of cellular injury
2.3: Understand different mechanisms of cell death
2.4: Identify systemic signs of cellular injury
Key Concepts
• Cellular adaptations may be adaptive or maladaptive.
• Hypoxia, reperfusion, and chemical agents may all result in free radical-induced damage.
• Irreversible cellular damage occurs when mitochondrial function and calcium homeostasis is lost.
• Cell death can be tightly regulated (apoptosis) or dysregulated (necrosis).
• Apoptotic cell death is often a normal aspect of tissue renewal.
• Cell damage and cell death result in both specific and non-specific biomarkers in tissues and in plasma.
Learning Units
Unit 2.2 Cellular Injury
Unit 2.3 Biomarkers of Cellular Injury
Unit 2.4 Cell Death
Unit 2.1 Cellular Adaptation


Figure 19: Cellular adaptation
Atrophy is in essence a decrease in cell size. There are multiple possible stimuli which can cause atrophy: nerve loss, malnutrition, aging, decreased workload. Perhaps not surprisingly, the actual mechanisms underlying atrophy are varied, and these can be physiologic or pathophysiologic. For example, muscle atrophy may occur as the result of either denervation or malnutrition, as depicted below (Figure 20).

Figure 20: Atrophy
In contrast, hypertrophy is an increase in cell size that is not due to increase in influx of H2O (cellular edema). A common pathological example is left ventricular hypertrophy, which is the result of the myocardium of the heart pumping against an elevated load. Hypertension, aortic stenosis, and diastolic overload are all causes of this type of cellular adaptation. Additionally, skeletal muscle hypertrophy occurs in individuals who engage in weight training (and/or take anabolic steroids). In both examples, the extracellular volume is primarily due to an increase in contractile proteins and mitochondria.
Hypertrophy can either be adaptive or maladaptive. Cardiac hypertrophy can increase cardiac output initially; however, increasing hypertrophy will eventually decrease compliance and reduce cardiac output. Eventually, an increase in cell size and tissue mass reduces the performance of any organ. In all cells at steady state, there is a balance between protein synthesis and degradation. During hypertrophy, the balance shifts towards protein synthesis. Although a variety of signaling pathways are involved in this shift in muscle cells, one of the most-studied involves the insulin-like growth factor (ILGF) pathway.
Whereas hypertrophy can involve tissues with limited mitotic ability (such as skeletal and cardiac muscle), hyperplasia can only occur in rapidly-dividing tissues, such as epithelial and glandular tissues, and in some cases smooth muscle cells. Hyperplasia involves an increase in cell number, and can also be adaptive or maladaptive. For example, in cases of liver damage or partial hepatectomy, compensatory hyperplasia of the liver can result in significant regeneration of the total organ mass and function. Hyperplasia can also be maladaptive: inappropriate endometrial cell mitosis and implantation outside the uterus occurs in endometriosis. Although the precise mechanisms are not well understood, both endocrine and immune dysfunctions have been implicated. Similarly, prostatic hyperplasia is maladaptive, and both examples have been implicated in progression to cancerous cell types.
Dysplasia is an abnormal change in cell size, shape, and organization, usually occurring in epithelial cells. It is not considered adaptive, but can be reversible (eg: cervical dysplasia). Dysplasia also occurs in pathologies of the gut, bone, respiratory epithelium, brain, and retina (Figure 21).

Figure 21: Dysplasia
Metaplasia involves the replacement of a mature cell type with another cell type (Figure 22). It often follows injury or insult, and occurs in a variety of tissue types. Metaplasia is considered an adaptation to external stimuli, such as bronchial and alveolar metaplasia in people exposed to cigarette smoke, and is not typically associated with increased risk of cancer. The mechanism(s) underlying metaplasia are complex, and are believed to involve reprogramming of undifferentiated stem cells.
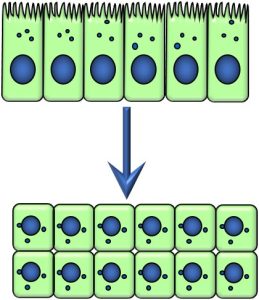
Figure 22: Metaplasia
Unit 2.2 Cellular Injury
Cells may be injured in a variety of ways, although for convenience we may consider four types or categories:
- Hypoxic injury (hallmark: loss of ATP)
- Free radical injury (hallmark: damage to biomolecules)
- Ischemia/reperfusion injury (hallmark: loss of antioxidant systems during initial hypoxic episode)
- Chemical (hallmark: loss of membrane integrity)
Hypoxic Injury
Hypoxia, or low oxygen tension in tissues, can be the result of ischemia (low blood flow) to tissues, or hypoxemia (low oxygen partial pressure in blood). As discussed in Chapter 1, oxygen is required to produce energy in the forms of ATP and NADH. Without adequate energy, molecular pumps such as the ubiquitous sodium/potassium ATPase cannot operate well enough to maintain low intracellular sodium levels (Figure 23). As sodium accumulates in the cytoplasm, water flows into the cell, as favoured by osmotic pressure. Organelles such mitochondria vacuolate, furthering hindering the production of cellular energy, and worsening the fluid and electrolyte imbalance within the cell. This ultimately may lead to cell death, when cellular integrity is lost (as discussed in Unit 2.4).
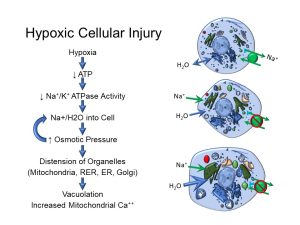
Figure 23: Hypoxic cell injury
Free Radical Injury
Before we can discuss free radical injury and its importance in many pathologies, it may be beneficial to review some basic chemistry. Recall that an atom consists of a nucleus comprising protons and neutrons, and an outer cloud of electrons. Electrons inhabit orbital shells which accommodate specific numbers of electrons. Electrons in the outermost (highest energy) orbital shell are referred to as valence electrons. In stable atoms, electrons in each orbital shell are usually paired. A free radical is an uncharged atom which has an unpaired valence electron (Figure 24):

Figure 24: Stable atoms vs. free radicals
Unpaired electrons make the atom unstable: they can give up or take away electrons from other atoms, similarly making them unstable. The process of losing electrons is called oxidation, whereas the process of gaining electrons is referred to reduction (Figure 25).
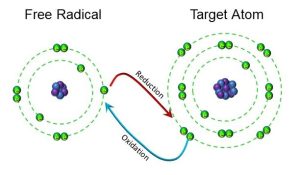
Figure 25: Oxidation and reduction
What makes free radicals particularly injurious to living systems is the fact that biomolecules such as lipids, nucleosides (including RNA and DNA), and proteins may all be damaged by free radicals, causing lipid peroxidation and loss of membrane integrity genetic damage and inactivation of enzymes, as depicted below (Figure 26).
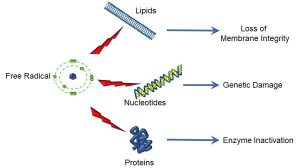
Figure 26: Targets of free radical damage
Several adult diseases have been either directly or indirectly associated with elevated free radicals: atherosclerosis, cancer, inflammatory joint diseases, asthma, diabetes, and cellular aging/senescence are but a few examples. Premature infants are even more at risk of free radical damage, owing to the immaturity of their antioxidant defense systems (discussed later within this unit), and diseases such as retinopathy of prematurity, bronchopulmonary dysplasia, and neurological disorders such as cerebral palsy have all been directly linked to free radical damage.
Effects of Oxidative State
In addition to causing damage to cellular structures, the relative balance of oxidants and reductants, or cellular redox status, greatly affects cellular activity, or cell state (Figure 27).
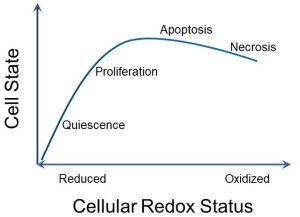
Figure 27: Cell state and cellular redox status
A key point is that a balance between oxidants and reductants is important in promoting cellular growth. In a highly reduced state, cellular growth stops (quiescence). A highly oxidized state promotes apoptosis and eventually necrosis (which we will discuss in Unit 2.4).
Cellular Antioxidant Mechanisms
Aerobic metabolism (Chapter 1) produces free radicals and reactive oxygen species (ROS) (Figure 28). Cells typically achieve balance in redox status via two mechanisms:
- Antioxidants, which block formation or scavenge free radicals (eg: Vitamins C and E, glutathione, transferrin)
- Enzymes, which convert free radicals into less injurious molecules (eg: superoxide dismutase (SOD), catalase, glutathione peroxidase)
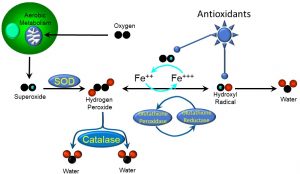
Figure 28: Antioxidant mechanisms
During aerobic metabolism, doublet oxygen is consumed, a byproduct of which is the potent superoxide radical. Endogenous antioxidant enzymes such as superoxide dismutase (SOD) dismute or transform the superoxide into hydrogen peroxide (H2O2), which is a reactive oxygen species. In the presence of ferrous iron (Fe++) and glutathione peroxidase, hydrogen peroxide easily transforms into the hydroxyl free radical (OH–·). Other free radical-scavenging enzymes such as catalase and glutathione reductase transform peroxides and hydroxyl free radicals, respectively, into less damaging molecules.
Regulation of Antioxidant Production
To more fully understand ischemia/reperfusion injury, which we will discuss next, it is important to appreciate how the body’s supply of antioxidants enzymes is regulated. Essentially, the whole system is similar to a ‘just in time’ business; that is, antioxidant enzymes are made as needed. Antioxidant genes contain regions which act as ‘sensors’ for redox status, called ‘antioxidant response elements’ (AREs). These are found in the promoter region of these genes and, when exposed to free radicals, regulatory proteins (such as Nuclear regulatory factor 2, or Nrf2) bind to and activate AREs. The end result is the transcription of genes which code for antioxidant proteins (Figure 29).
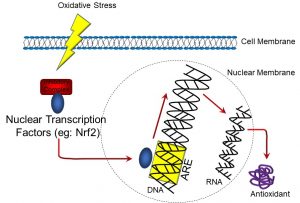
Figure 29: Oxidative stress and antioxidant response elements
Conversely, when oxidative stress decreases, such genes are downregulated. This saves the cell from expending energy for producing antioxidant enzymes when they are not needed. However, this is a condition that is central to ischemia-reperfusion injury.
Ischemia-Reperfusion Injury
Much of our previous discussion is necessary for a more fulsome understanding of ischemia/reperfusion injury. Tissues and organs which have reduced blood flow (ischemia) also experience low oxygen tension (hypoxia). Conditions which can transiently cause ischemia include various types of strokes, coronary artery insufficiencies, and transplants. Localized hypoxia in turn decreases (downregulates) the expression of antioxidant enzymes.
When circulation, and therefore oxygenation, are restored, reactive oxygen species such as hydroxyl and superoxide free radicals and hydrogen peroxide are again produced. However, initially there will be a deficit in antioxidant enzymes, resulting in a redox imbalance or pro-oxidant state. Though antioxidant enzyme production can be upregulated, as discussed previously, in the meantime the aforementioned free radicals and reactive oxygen species can cause cellular damage.
Chemical Injury
There are innumerable types of chemicals which can cause cellular injury. Examples of the more common agents (and their modes of toxicity) include:
- Cigarette smoke
o Contain polyaromatric hydrocarbons (PAHs), which metabolize to form ‘adducts’ or compounds that bind to DNA, as well as forming free radicals (linked to several forms of cancer)
o PAHs also bind to estrogen receptors - Lead
o Inactivate enzymes, displaces Ca++ in bone
o Causes acute liver damage, disrupts neuronal signalling - Organic Solvents
o Include alcohols, toluene, carbon tetrachloride
o Damage lipid membranes, metabolized to free radicals
o Form toxic metabolites, such as aldehydes (alcohol) - Carbon monoxide
o Out-competes oxygen for binding sites on hemoglobin
Radiation Injury
While there are many different forms of radiation, we will refer herein to ‘ionizing radiation’. This consists of any type of subatomic particle or electromagnetic wave that has sufficient energy to remove electrons or ionize atoms. The primary types of ionizing radiation are:
- X-rays and gamma-rays (consist of photons)
- Alpha particles (consist of two protons and two neutrons)
- Beta particles (essentially electrons)
In general, radiation can cause instability in any biomolecule, although some of the main cellular effects can include:
- DNA strand breaks and misparings, causing genomic instability and increased rates of mutation
- Generation of free radicals and chemical damage
- Induction of apoptosis and/or necrosis
Self-Study Question:
Unit 2.3 Biomarkers of Cellular Injury

Tissue Signs of Cellular Injury
Markers or signs of cell damage and/or cell death can be classified as follows:
- Endogenous products found in elevated levels in tissues (eg: pathogenic calcification). Injured cells accumulate calcium in atherosclerotic lesions, termed ‘dystrophic’ calcium. Calcium deposits in tissues are also evident in certain cancers, in which dead and dying cells leave ‘metastatic’ calcifications (Figure 30). Certain conditions result in elevated calcium levels, such as hyperparathyroidism, immobilization, or cancer. This is due to either excessive production, or ineffective catabolism and/or excretion.
.jpg)
Figure 30: Metastatic calcifications. Pale, tan areas (arrows) represent deposits of calcium from dead and dying cells, the result of metastatic lung cancer.
- Endogenous substance that accumulates within cells. For example, Tay-Sachs disease is a neurodegenerative disorder in which individuals lack an enzyme called hexosaminidase. This results in a build-up of fatty material in lysosomes.
- Accumulation of exogenous products (eg: carbon from coal dust)
Self-Study Question:
What is ‘fatty liver disease’?
Plasma Markers of Cellular Injury
Cell/tissue/organ damage often releases endogenous substances which end up in plasma or serum. Clinical laboratory tests include many of these markers of injury, as depicted in the sample laboratory requisition below (Figure 31).

Figure 31: Plasma markers of cellular injury
The more clinically-relevant markers include:
- Lactate dehydrogenase (LDH): a very non-specific marker
- Alanine aminotransferase (ALT) and aspartate aminotransferase (AST): relatively specific for hepatocellular injury
- Creatine kinase (CK): a marker of cardiac injury
- Uric acid: elevated in gouty arthritis
- Amylase and lipase: both are products of the exocrine pancreas and can be elevated as a result of pancreatitis
Urinary and Fecal Markers of Cellular Injury
Laboratory testing of urine and feces is often done to assess markers of cell and tissue damage. Some of the major biomarkers found in these substances are:
- Microalbuminuria: albumen is typically not filtered or secreted into the nephron; indicator of glomerular damage
- Creatinine: a muscle waste product; elevated levels can indicate muscle damage (rhabdomyolysis)
- Extracellular vesicles: released during renal tubular necrosis
- Prostate-specific antigen: a marker of prostatic hyperplasia
- Uric acid: a waste product of protein metabolism; elevated in urine as a result of gouty arthritis
- 8-OHdG (8 hydroxy-deoxy guanine): an oxidized purine nucleobase that is an indicator of free radical-induced DNA damage
- Calprotectin: a product of neutrophils; one indicator of intestinal inflammation
Self-Study Question:
Can you identify other markers of cell and tissue damage?
Unit 2.4 Cell Death

Cellular damage eventually increases to the point where cellular function and integrity are lost. Typically this occurs when mitochondrial function ceases, intracellular calcium is dysregulated, and cell membrane integrity is lost. Cell death may be the result of injury that is nonrecoverable, or a normal developmental process. Microscopically, cell death is not necessarily easy to detect, although there are a variety of sensitive assays which may be used for these purposes. For example, in the microscope image below, live and dead cells are distinguishable on the basis of selective fluorescent dyes (Figure 32).
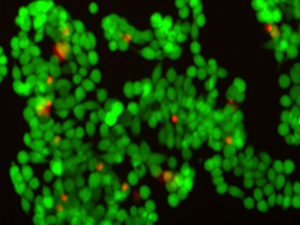
Figure 32: Live vs. dead cells, visualized with fluorescent markers
In this image taken with a specialized microscope, we see a collection of colon cancer cells. The red dots are in fact nuclei in dead cells which, due to a loss of plasma membrane integrity, stain with a red fluorescent dye (ethidium). The green dots are living cells which take up a membrane-permeant dye (calcein).
Within any tissue or organ, countless cells are in the process of dying, although it is the balance between growth and death which reflects many disease states. Moreover, what the micrograph above does not tell us is the specific type of cell death that is occurring, of which we can delineate three: necrosis, apoptosis, and autophagy. Some of the principle features of these different types of cell death are depicted below (Figure 33).
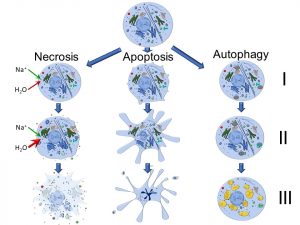
Figure 33: Necrosis, apoptosis, and autophagy
Self-Study Question:
How might cell death help to maintain homeostasis?
Necrosis
Necrosis is fundamentally a loss of plasma membrane integrity in which cellular components are released in an indiscriminate manner. This is characterized by cellular and organellar swelling, and rupture of the cell. We can identify four discrete stages of this process:
- Loss of ATP-driven pumps; influx of sodium and water
- Cytosolic and organelle swelling; leakage of lysosomes and proteasomes
- Loss of intracellular and extracellular membrane integrity; and
- Release of organellar and cytosolic contents into intracellular space
At the macroscopic level, necrosis can be classified as ‘liquefactive’ or ‘coagulative’. In liquefactive necrosis, there is partial or total dissolution of dead tissue, resulting in a liquid mass. Hydrolytic enzymes, released by either lysosomes or bacteria, are responsible for liquefactive necrosis, which is more commonly associated with bacterial infections. Coagulative necrosis is characterized by protein cross-linking and the maintenance of relatively normal tissue and cellular architecture. Ischemia or hypoxia are common causes of coagulative necrosis in tissues, with the exception of the brain. Other sub-categories of necrosis include caseous necrosis (seen in tuberculosis), fat necrosis (seen in fatty tissues in which lipids break down to form free fatty acids), fibrinoid necrosis (associated with vascular damage in which there are excessive fibrin deposits), and gangrenous necrosis (a form of ischemia-provoked necrosis occurring in limbs or digits). Conventionally, necrosis has been viewed as a non-regulated process, although there is evidence that necrosis can be genetically programmed. Necroptosis is a well-known viral defense mechanism, and is associated with conditions such as Crohn’s disease and pancreatitis.
Apoptosis
Apoptosis comes from the Greek term ‘ptosis’, which translates to ‘drooping’ or ‘falling off’. Several discrete stages of apoptosis have been identified, which include:
I. Membrane blebbing, increased mitochondrial membrane permeability, and activation of caspase pathways
II. Nuclear compaction (pyknosis) and cellular shrinkage
III. Nuclear fragmentation (karyorrhesis) and formation of apoptotic bodies
One of the more visible microscopic changes is pyknosis (Figure 34):
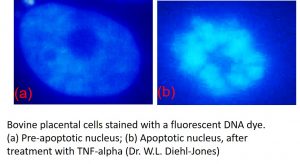
Figure 34: Pyknosis. Bovine placental cells stained with a fluorescent DNA dye. (a) Pre-apoptotic nucleus; (b) Apoptotic nucleus, after treatment with TNF-alpha (Image by Dr. W.L. Diehl-Jones)
We can consider both extrinsic and intrinsic apoptosis pathways. Intrinsic pathways are activated as part of developmental processes and via external signals such as ionizing radiation, growth factor depletion, or hormones such as glucocorticoids. In this regard, apoptosis may be considered a normal part of cellular turnover, in which skin, gut and other epithelial cells are shed via apoptosis as new cells develop. Extrinsic pathways can be activated by specific extracellular ligands released by other cells. In both cases, a series of enzymes called caspases are activated, which then kill the cell by causing protein degradation. Several genes, including BNIP3 and Nix, have been identified as regulators of apoptosis. A key component of apoptotic cell death is a loss of mitochondrial membrane potential and dysregulated intracellular calcium. ‘Faulty’ apoptosis – that is, apoptosis pathways which are disrupted – may result in defective cells surviving and passing on defective characteristics and/or resulting in tumourigenesis.
Autophagy
Essentially autophagy is a regulated form of ‘self eating’. Causes of autophagy include starvation and other toxic insults which trigger the cell to deliver cell components for digestion by lysosomes. A series of ‘autophagy-related genes’ (ATGs) are expressed during times of cellular stress, and activates the formation and delivery of autophagosomes to lysosomes. This promotes cell survival by recycling essential components and conserves cellular resources. However, autophagy can also activate apoptosis or necrosis. Autophagy can also mediate developmental processes. Three hallmarks of autophagy include:
I. Formation of autophagosomes
II. Fusion of autophagosomes and lysosomes
III. Destruction of intracellular organelles
CHAPTER SUMMARY
When the environment changes, cells and tissues either adapt or die. Some forms of adaptation are beneficial, while others are not, resulting in disease. Moreover, cell death may take different forms, and may in fact be a normal part of cell and tissue physiology. Going forward in other pathophysiology courses, an appreciation of the cellular adaptation and death will help you to have a more fulsome understanding of many different disease states.
proteins with a sequence similarity to insulin; part of a complex signalling system between cells and the environment.
inadequate blood supply to an organ or part of the body
