76
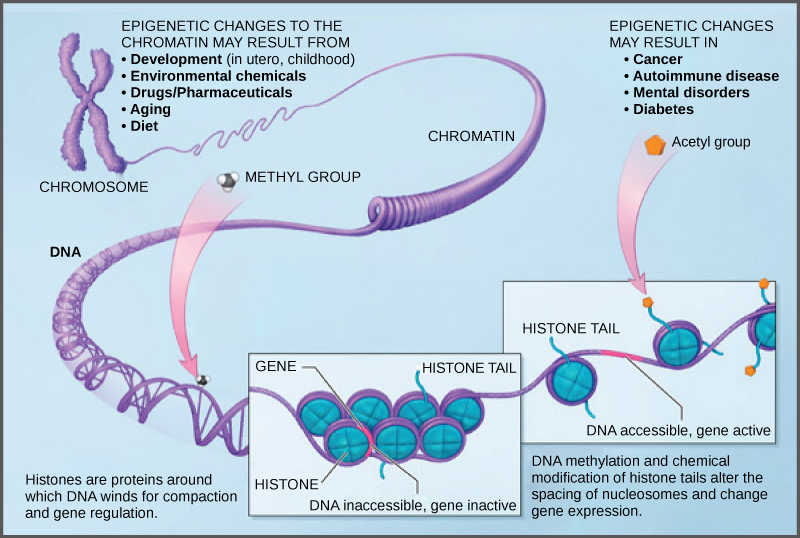
DNA modifications that do not change the DNA sequence can affect gene activity. Chemical compounds that are added to single genes can regulate their activity; these modifications are known as epigeneticchanges. The epigenomecomprises all of the chemical compounds that have been added to the entirety of one’s DNA (genome) as a way to regulate the activity (expression) of all the genes within the genome. The chemical compounds of the epigenome are not part of the DNA sequence, but are on or attached to DNA (“epi-“ means above in Greek). Epigenomic modifications remain as cells divide and in some cases can be inherited through the generations. Environmental influences, such as a person’s diet and exposure to pollutants, can also impact the epigenome.
Epigenetic changes can help determine whether genes are turned on or off and can influence the production of proteins in certain cells, ensuring that only necessary proteins are produced. For example, proteins that promote bone growth are not produced in muscle cells. Patterns of epigenome modification vary among individuals, different tissues within an individual, and even different cells.
Regulating Access to Genes
The human genome encodes over 20,000 genes, which means that each of the 23 pairs of human chromosomes contains thousands of genes. The DNA in the nucleus of each cell is precisely wound, folded, and compacted into chromosomes so that it will fit inside the nuclear membrane. It is also organized so that specific segments can be accessed as needed by a specific cell type.
The first level of organization, or packing, is the winding of DNA strands around histoneproteins. Histones package and order DNA into structural units called nucleosomecomplexes, which can control the access of proteins to the DNA regions (Figure 1a). Under the electron microscope, this winding of DNA around histone proteins to form nucleosomes looks like small beads on a string (Figure 1b). These beads (histone proteins) can move along the string (DNA) and change the structure of the molecule.
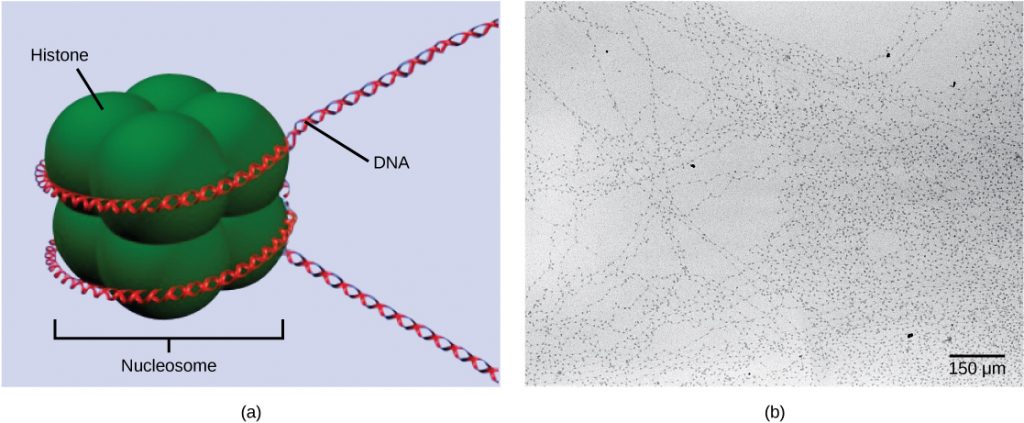
If DNA encoding a specific gene is to be transcribed into RNA, the nucleosomes surrounding that region of DNA can slide down the DNA to open that specific chromosomal region and allow for the transcriptional machinery (RNA polymerase) to initiate transcription. Nucleosomes can move to open the chromosome structure to expose a segment of DNA, but do so in a very controlled manner.
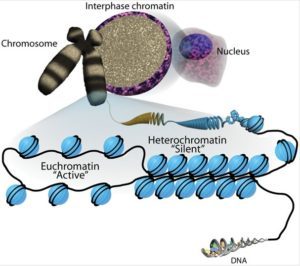
Active open regions of chromatin are called euchromatin (Figure 2).Regions of the genome that are transcriptionally active are typically euchromatic. Tightly wound regions of chromatin are called heterochromatin.Heterochromatic regions of the genome are typically silenced and transcriptionally inactive.
Modifications to DNA and histones
How the histone proteins move, and whether the DNA is wrapped loosely or tightly around them, is dependent on signals found on both the histone proteins and on the DNA. These signals are chemical tags added to histone proteins and DNA that tell the histones if a chromosomal region should be open or closed. These tags are not permanent, but may be added or removed as needed. They are chemical modifications (phosphate, methyl, or acetyl groups) that are attached to specific amino acids in the protein or to the nucleotides of the DNA. The tags do not alter the DNA base sequence, but they do alter how tightly wound the DNA is around the histone proteins.
This type of gene regulation is called epigenetic regulation. Epigenetic means “around or above genetics.” The changes that occur to the histone proteins and DNA do not alter the nucleotide sequence and are not permanent. Instead, these changes are temporary, although they can and often do persist through multiple rounds of cell division. They alter the chromosomal structure (open euchromatin or closed heterochromatin) as needed, but do not change the sequence of bases within the DNA.
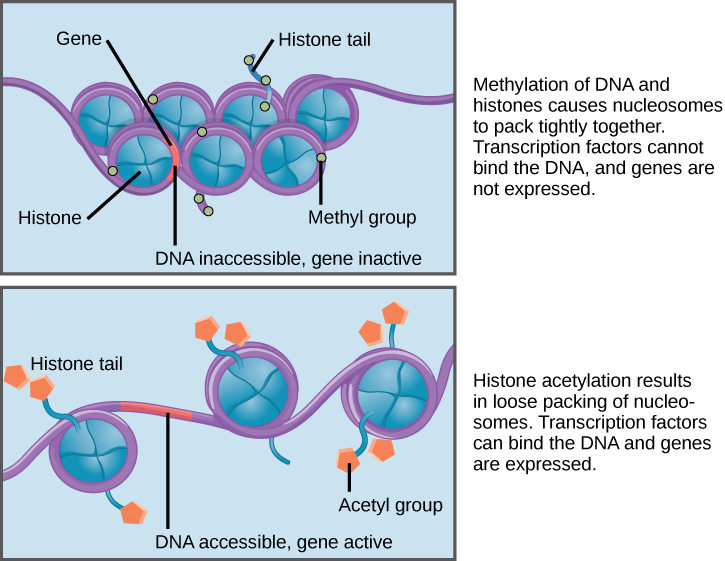
A gene can be turned on or off depending upon the location and modifications to the histone proteins and DNA. If a gene is to be transcribed, the histone proteins and DNA are modified surrounding the chromosomal region encoding that gene. This opens the chromosomal region (it becomes euchromatic) to allow access for RNA polymerase and other proteins, called transcription factors, to bind to the promoter region, located just upstream of the gene, and initiate transcription. If a gene is to remain turned off, or silenced, the histone proteins and DNA have different modifications that signal a closed chromosomal configuration. In this closed configuration (heterochromatin), the RNA polymerase and transcription factors do not have access to the DNA and transcription cannot occur (Figure 2).
DNA Methylation
A common type of epigenomic modification is called methylation.Methylation involves attaching small molecules called methyl groups, each consisting of one carbon atom and three hydrogen atoms, to DNA nucleotides or the amino acids that make up the histone proteins.
When DNA is methylated, the methyl group is typically added to cytosine nucleotides. This occurs within very specific regions called CpG islands. These are stretches with a high frequency of cytosine and guanine dinucleotide DNA pairs (CG) found in the promoter regions of genes. When this configuration exists, the cytosine member of the pair can be methylated (a methyl group is added). This modification changes how the DNA interacts with proteins, including the histone proteins that control access to the region. When methyl groups are added to a particular gene, that gene is turned off or silenced, and no protein is produced from that gene (Figure 3).
“Histone Code” Hypothesis
The histone code hypothesisis the hypothesis that transcription of a gene is in part regulated by modifications made to histone proteins, primarily on their somewhat floppy ends (their “tails”). Many of the histone tail modifications correlate very well to chromatin structure and both histone modification state and chromatin structure correlate well to gene expression levels. The most important concept in the histone code hypothesis is that the histone modifications serve to recruit other proteins by specific recognition of the modified histone, rather than through simply stabilizing or destabilizing the interaction between histone and the underlying DNA. These recruited proteins then act to alter chromatin structure actively or to promote transcription.
The histone code has the potential to be massively complex. There are at least 20 modifications that are made to histone tails that have been relatively well characterized, and there is the potential for many more that we have not discovered. Each histone can be modified on multiple amino acids, with multiple different chemical modifications. The information that can be stored in the histone code dwarfs the amount that is stored in the order of the bases in the human genome.
Histone Methlyation
A portion of the histone protein known as the histone tail can have methyl groups (CH3) added to it. This is the same modification that is made to cytosine nucleotides in DNA. The specific amino acid in the histone tail that gets methylated is very important for determining whether it will tighten or loosen chromatin structure. Modification to several amino acids in the tail is correlated with euchromatin and active transcription, while modification to other amino acids is correlated with heterochromatin and gene silencing. You should know that histones can be methylated, but we can’t use histone methylation as a predictor for euchromatin or heterochromatin.
Histone Acetylation
Histone tails can also be modified by the addition of an acetyl group (this process is known as acetylation). If you remember from cellular respiration, an acetyl group (such as that found in acetyl-CoA) is a 2-carbon molecule. When histone tails are acetylated, this typically causes the tails to loosen from around the DNA, allowing the chromatin to loosen (Figure 3).
Other modifications
There are many other modifications that can be made to histone proteins in addition to methylation and acetylation. Histone tails can be phosphorylated or ubiquitinated (where a small protein called ubiquitin is attached). Histone phosphorylation seems to be related to DNA repair. Ubiquitination has been shown to be associated with both transcriptional activation or inactivation, depending on the specific location.
Epigenetic Changes
Because errors in the epigenetic process, such as modifying the wrong gene or failing to add a compound to a gene, can lead to abnormal gene activity or inactivity, they can cause genetic disorders. Conditions including cancers, metabolic disorders, and degenerative disorders have all been found to be related to epigenetic errors.
Cancerous cells often have regions of DNA that show different levels of methylation compared to normal cells. Some genes are methylated and silenced in cancerous cells, while they are unmethylated and active in normal cells. Other genes are active in cancerous cells, but inactive in normal cells. Each specific cancer in each specific individual can show different patterns of methylation, although there are similarities between many different types of cancer.
Scientists continue to explore the relationship between the genome and the chemical compounds that modify it. In particular, they are studying what effect the modifications have on gene function, protein production, and human health.
References
Unless otherwise noted, images on this page are licensed under CC-BY 4.0 by OpenStax.
OpenStax, Concepts of Biology. OpenStax CNX. May 18, 2016 http://cnx.org/contents/b3c1e1d2-839c-42b0-a314-e119a8aafbdd@9.10
