BINDEVEVSSYKDOMMER (REV 021-033)
54 Antisyntetasesyndrom (ASS) (REV 021, REV 029)
Øyvind Palm and Jan Tore Gran
Diagnosekoder ICD-10: M35.8 + J99.1 Prosedyrekoder: 6-minutter gangtest: FYFX05. Kapillærmikroskopi: PKFT00. Intravenøs infusjon: WBGM00. Infusjon med gammaglobulin: RPGM05. EKG: FPFE15 ATC koder (for legemiddelstatistikk): L04A A Immunsuppressive legemidler. Behandling med rituksimab: L01XC02. Behandling med cyklofosfamid: L01AA01.
Nøkkelord for journalskriving
- Interstitiell lungesykdom (ILD)
- CT med typisk mattglass i begge lungers nedre områder.
- Myositt (poly- eller dermatomyositt)
- CK, LD, ASAT forhøyet
- MR av lårmuskler
- Dynamisk røntgen av øsofagus (dysfagi)
- Muskelbiopsi
- Dermatomyositt-forandringer (Gottrons, V- eller Sjal-tegn, helioptropt)
- Artritt
- Raynauds fenomen
- Svelgevansker
- Mekanikerhender (sprukne fingertupper)
- Feber
- Antistoff: Jo-1, PL-7*, PL-12*, andre (OJ, EJ, KS, Zo)*
*myositt-spesifikke antistoff ved myositt-blot test/
Hensikten med konsultasjonen.Innhold
- Definisjon
- Historikk
- Patogenese
- Epidemiologi
- Symptomer
- Utredning
- Klassifikasjonskriterier
- Differensialdiagnoser
- Svangerskap ved ASS
- Behandling
- Prognose
- Retningslinjer og prosedyrer
- Litteratur
Definisjon
Antisyntetasesyndromet (ASS) er en sjelden, autoimmun bindevevssykdom bestående av en variabel kombinasjon av v interstitiell lungesykdom (ILD), myositt, og leddmanifestasjoner som artralgi eller artritt. I tidlige sykdomsstadier kan uspesifikke symptomer som feber og kutane manifestasjoner dominere det kliniske bildet, før mer typiske organmanifestasjoner utvikles. Lungemanifestasjoner er særlig sentrale, med en inflammasjon som kan variere fra mild til alvorlig og som ofte viser progresjon over tid (Wells M, 2022).
Syndromet har fått sitt navn fra den karakteristiske gruppen av autoantistoffer mot aminoacyl-tRNA-syntetaser som alltid påvises. Disse enzymene som angripes er essensielle for proteinsyntesen. Autoantistoffene antas derfor å være patogenetisk relevante, og de danner grunnlaget for diagnostiske kriterier.
De vanligste antistoffene er anti-Jo-1 (påvist hos 50–80 % av tilfellene), anti-PL-7 (12–20 %) og anti-PL-12 (12–20 %). Andre ASS-assosierte autoantistoffer forekommer sjeldnere, men anti-SSA (Ro52 eller Ro60) ses hos omtrent 50 %, vanligvis i lave til moderate titere (Wells M, 2022; Cavagna L, 2015).
Historikk
Antisyntetasesyndromet ble første gang beskrevet på 1980-tallet som en distinkt klinisk fenotype innenfor idiopatisk inflammatorisk myopati. Etter identifikasjonen av anti-Jo-1 som det første og hyppigste antistoffet i gruppen, ble det gradvis klart at pasienter med disse autoantistoffene delte en karakteristisk kombinasjon av myositt, interstitiell lungesykdom og ekstra-muskulære manifestasjoner (Margurie C, 1990).
Senere ble flere andre aminosyl-tRNA-syntetaseantistoffer identifisert, deriblant anti-PL-7, anti-PL-12, anti-EJ, anti-OJ og andre sjeldnere varianter, noe som bidro til å etablere ASS som et heterogent, men veldefinert klinisk syndrom. Den økende forståelsen av sykdommens immunologiske bakgrunn har siden gitt grunnlag for dagens diagnostiske kriterier og for utviklingen av mer målrettet behandling.
Patogenese
Patogenesen ved ASS er kompleks. Tilgjengelig forskning peker på et samspill mellom genetisk disposisjon, miljøfaktorer og immunologiske mekanismer som fører til tap av immunologisk toleranse og utvikling av autoimmunitet.
1. Autoimmun aktivering og tap av toleranse. Den utløsende årsaken (etiologien) er ukjent, men som ved andre autoimmune sykdommer, foreligger det en svikt i immunsystemets evne til å tolerere egne proteiner og vev (autoimmunitet). Sentralt i ASS er immune responser rettet mot aminoacyl-tRNA-syntetaser, enzymer som er essensielle i proteinsyntesen. Det antas at vevsskade kan frigjøre aminoacyl-tRNA og syntetaseproteiner fra skadede celler, noe som igjen kan stimulere antigenpresentasjon og utløse en kaskade av immunologiske reaksjoner.
2. Autoantistoffer mot aminoacyl-tRNA-syntetaser. ASS kjennetegnes av antistoffer rettet mot spesifikke syntetaser, blant annet anti–Jo-1, anti–PL-7, anti–PL-12 og flere andre. Hver aminosyre har sin egen tRNA-syntetase, og ASS-relaterte antistoffer er spesifikke for den enkelte syntetase. En direkte patogenetisk rolle for disse antistoffene er imidlertid ikke definitivt påvist, men de fungerer som viktige immunologiske markører som gjenspeiler underliggende autoimmun aktivering.
3. Lungene som mulig primærarena for sykdomsutvikling. Flere funn tyder på at sykdommen begynner i lungevevet. CD4⁺ T-celler som er reaktive mot aminoacyl-tRNA og anti–Jo-1 antistoff er identifisert i bronkialvæske hos pasienter med ASS (Galindo-Feria AS, 2020). Dette styrker hypotesen om at lungeinflammasjon, antigeneksponering eller ekstern irritasjon via luftveiene kan være et tidlig sykdomsdrivende moment. Den hyppige forekomsten av interstitiell lungesykdom ved ASS understøtter også denne modellen.
, CC BY-NC-SA 3.0")
4. Kronisk immunaktivering og vevsskade. Når sykdommen er etablert, vedlikeholdes den av:
- CD4+ T-celler som aktiverer B-celler til antistoffproduksjon.
- CD8+ cytotoksiske T-celler som angriper muskelceller og alveolært epitel.
- Makrofaginfiltrasjon i muskler (perimysial dominans) og i lungevev.
- Komplementmediert cytotoksisitet.
- Fibrose, særlig i lunge og muskel, som følge av kronisk inflammasjon.
5. Genetisk predisposisjon. Genetiske faktorer kan medføre sårbarhet for miljøpåvirkningspiller. Variasjonen HLA-DRB1*03:01, som er assosiert med økt risiko for flere autoimmune sykdommer, er funnet hos rundt 73 % av pasienter med ASS, sammenlignet med 23 % blant friske kontroller (O’Hanlon TP, 2006). En polymorfisme av IL1B-genet (rs1143634) som koder for det proinflammatoriske cytokinet IL-1, er funnet å være assosiert med økt risiko for ASS. GA-genotypen av denne polymorfismen er også knyttet til økte serumnivåer av IL-1 hos ASS-pasienter. Allelet MUC1 rs4072037 er også assosiert med økt risiko for ASS. I motsetning til andre interstitielle lungesykdommer (ILD), har ASS-ILD ikke vist en sammenheng med MUC5B-genpolymorfismen (Patel P, 2024).
6. Miljøfaktorer som triggere. Miljømessig eksponering antas å bidra både til sykdomsinitiering og sykdomsforverring. Irritanter og antigener som inhaleres, inkludert tobakksrøyk, vaskemiddelpartikler, fugleekskrementer og luftforurensning kan indusere inflammatoriske responser i luftveiene. Dette kan føre til vevsskade, lokal frigjøring av autoantigener og aktivering av både innate og adaptive immunresponser og dermed øke risikoen for å trigge autoimmun sykdom hos genetisk predisponerte individer.
")
Epidemiologi
Forekomsten av ASS i den generelle befolkningen er ikke godt kartlagt.
Prevalens: Orphanet (per oppdatering 16. oktober, 2025) anslår prevalensen til 1-9 per 100 000.
Insidens. En studie i indre byområder av Manchester avdekket en gjennomsnittlig insidens av IIM (Idiopatisk inflammatorisk myopati, definert av EULAR/ACR-klassifikasjonskriteriene) på 17,6 per 1 000 000 personår. Av disse ble 28% identifisert som ASyS ved ekspertkonsensus (Parker MJS, 2018). Det tilsvarer en årlig insidens på ca. 5 per million.
Kvinner rammes oftere enn menn, og gjennomsnittsalderen ved debut er ca. 50 år (ofte mellom 43 og 60 år) (Legout L, 2002). Svarte pasienter kan ha mer hyppig og alvorlig ILD (interstitiell lungesykdom), men ASyS-epidemiologien ser ellers ikke ut til å være påvirket av etnisitet (Pinal-Fernandez I, 2017; Chua F, 2012).
Symptomer ved ASS
Antisyntetasesyndrom (ASS) er en heterogen tilstand med varierende klinisk presentasjon. Sykdomsdebuten kan være akutt, subakutt eller snikende, og kombinasjonen av symptomer varierer betydelig mellom pasienter. Typiske manifestasjoner inkluderer systemiske symptomer, myositt, interstitiell lungesykdom (ILD), hudforandringer, artritt og i enkelte tilfeller malignitetstilstander.
1. Systemiske symptomer. Sykdommen debuterer ofte med generell inflammasjon og påvirket allmenntilstand. Vanlige symptomer er feber, nattesvette og vekttap, noe som kan gi et inntrykk av infeksjon eller malignitet. Samtidig kan enkelte pasienter presenterer ha et mer snikende forløp og kan være nesten asymptomatiske i starten.
2. Myositt. ASS er klassifisert som en inflammasjonsmyopati, men graden av muskelinvolvering varierer. Omtrent 25 % av pasientene har hypomyopatisk eller amyopatisk sykdom, det vil si få eller ingen kliniske tegn til muskelsvakhet. Kreatinkinase (CK) kan være moderat forhøyet eller normal (Cavagna L, 2017: Wells M, 2022). Når myositt foreligger, foreligger vanligvis proksimal muskelsvakhet, myalgi og redusert utholdenhet.
3. Lungesymptomer og Interstitiell lungesykdom (ILD). Interstitiell lungesykdom er en av hovedmanifestasjonene og anses som den mest alvorlige komponenten ved ASS, samtidig som den er den vanligste dødsårsaken. ILD kan utvikle seg raskt eller gradvis, og både radiologiske funn og lungefunksjonstester viser ofte mer alvorlig sykdom enn ved andre former for myositt.
Klinisk rapporterer omtrent 50 % gradvis økende dyspné og tørrhoste, mens den andre halvparten får akutt respiratorisk forverring (Tillie-Leblond I, 2008). Før antistoffer påvises, kan presentasjonen ligne hypersensitivitetspneumoni (Kawano-Dourado L, 2013). ILD-mønstre inkluderer typisk NSIP eller OP, men overlapper er hyppige.
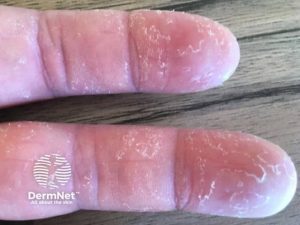
4. Hudmanifestasjoner. Hudforandringer forekommer hos 16–21 % av pasientene og kan være diagnostisk viktige. Den mest karakteristiske manifestasjonen er “mekanikerhender” som innebærer fissurerte, hyperkeratotiske og erytematøse forandringer særlig på de laterale sidene av fingre og tommel (Hervier B, 2013). Håndflater og håndrygger affiseres vanligvis ikke. Tilsvarende hyperkeratose på tær omtales som “hiker’s feet” (Trallero-Araguas E, 2016).
Omtrent 20 % har hudforandringer mer typiske for dermatomyositt, og over 50 % har Raynauds fenomen.
5. Leddmanifestasjoner. Artralgi og artritt er vanlige og ses hos mer enn 50 % av pasientene. Artrittbildet er typisk symmetrisk, polyartikulært og non-erosivt. Det er vanligere blant pasienter med anti–Jo-1 antistoff (Cavagna L 2019). Likevel kan erosiv artritt forekomme hos 15–20 % av pasientene.
Tidlig polyartritt er ofte assosiert med revmafaktorer (RF) og anti-CCP-antistoffer, noe som kan gi et klinisk bilde som overlapper med revmatoid artritt (Gonzalez-Gay MA, 2018); (Cavagna L, 2010).
Malignitet. Selv om dermatomyositt generelt er assosiert med økt kreftrisiko hos voksne, viser større kohortstudier ingen økt forekomst av malignitet ved ASS sammenlignet med befolkningen ellers (Pinal-Fernandez I, 2017). Likevel anbefales malignitetsscreening dersom pasienten presenterer dermatomyositt-lignende symptomer eller andre forhold som gir mistanke om underliggende kreftsykdom.
Utredning
Utredningen ved ASS krever en systematisk vurdering av symptomer, kliniske funn, laboratorieprøver, bildediagnostikk og eventuelt muskelbiopsi. Sykdomsmanifestasjonene varierer betydelig, og både muskler, lunger, hud og ledd må vurderes.
Anamnesen Anamnesen bør fokusere på symptomer relatert til myositt, interstitiell lungesykdom (ILD), hudforandringer og leddaffeksjon. De viktigste spørsmålene omfatter:
-Muskler: Spør om pasienten opplever svakhet, spesielt ved å reise seg fra en stol, å gå i trapper, å løfte armene over hodet og dysfagi (øsofagus-manifestasjon).
-Lunger: Tung pust ved anstrengelse og tørrhoste er typiske symptomer. ILD kan debutere gradvis eller akutt, og er ofte mer alvorlig enn ved andre myositter (Tillie-Leblond I, 2008).
-Hud: “Mekanikerhender” (hyperkeratose og fissurer lateralt på fingrene) er karakteristisk, samt Raynauds fenomen.
-Ledd: Spør etter smerte, hevelse og stivhet, særlig i småledd i hender og føtter. Polyartritt forekommer hyppigere hos pasienter med anti-Jo-1 (Cavagna L 2019).
Klinisk undersøkelse. Undersøkelsen bør inkludere generell status med vekt på muskelstyrke, lungeauskultasjon, hudfunn og ledd.
- Myositt: Ikke alltid klinisk påvisbar. Observer funksjonelle tester som evne til å reise seg fra stol eller fra huksittende.
- ILD: Krepitasjoner basalt ved auskultasjon kan være første tegn.
- Hud: Mekanikerhender og “hiker’s feet” vurderes (Hervier B, 2013; Trallero-Araguas E, 2016).
- Raynauds fenomen: Vanlig, men ofte mildere enn ved systemisk sklerose eller MCTD.
- Artritt: Kan være ikke-erosiv eller erosiv (15–20 %) (Cavagna L 2019).
- Sicca-symptomer: Kan tyde på overlapp med Sjøgrens syndrom.
Laboratorieprøver kan omfatte CRP, SR, Hb, leukocytter med differensialtelling, trombocytter, lever-, nyre-, og thyreoidea-funksjonsprøver, elektrolytter, kreatinin kreatin kinase (CK), albumin, glukose. Troponin ved mistanke om myositt. Urin-stiks. I tillegg tas antistoff-prøver som kan omfatte “myositt-spesifikke prøver” og myositt-relaterte prøver (se tabellen nedenfor).
Tabell: Laboratorieprøver ved antisyntetasesyndromet (ASS)
| Prøvetype | Parameter | Relevans / typiske funn | Kommentar / Referanser |
|---|---|---|---|
| Inflammasjonsmarkører | CRP, SR | Kan være normale eller forhøyede | Ikke spesifikke, men øker ved aktiv inflammasjon |
| Hematologi | Hb, leukocytter m/diff., trombocytter | Anemi ved kronisk inflammasjon, leukocytose eller leukopeni mulig | Del av generell systemvurdering |
| Biokjemi | Elektrolytter, kreatinin, leverprøver, albumin, glukose | Vurderer organfunksjon, ernæringsstatus og differensialdiagnoser | Relevante ved multisystemisk sykdom |
| Thyreoideaprøver | TSH, fT4 | Utreder samtidig autoimmun thyreoideasykdom | ASS kan overlappe andre autoimmune tilstander |
| Muskelenzymer | CK | Normal eller moderat forhøyet selv ved myositt | Hypo/amyopatiske forløp er vanlige (Cavagna L, 2017; Wells M, 2022) |
| Troponin I | Tas ved mistanke om myokardaffeksjon | Skiller mellom myokardial og skjelettmuskelskade | |
| Urin | Urin-stiks | Proteinuri/hematuri ved diferensialdiagnoser | Screening for nyreaffeksjon |
| Myosittspesifikke antistoffer (MSA) | Anti–Jo-1 | Vanligst (50–80 %). Klassisk ASS med ILD (>80 %), myositt, hudforandringer og artritt (~70 %) | Spesifikt for histidyl–tRNA-syntetase |
| Anti–PL-7 | 12–20 %. Alvorlig ILD, mild myositt, Raynaud | Threonyl–tRNA-syntetase | |
| Anti–PL-12 | 12–20 %. Alvorlig ILD, mild myositt, Raynaud | Alanyl–tRNA-syntetase | |
| Anti–OJ | 5–10 %. Mest lungemanifestasjoner, sjeldnere myositt, artritt kan forekomme | Isoleucyl–tRNA-syntetase | |
| Anti–EJ | 4–20 %. DM-lignende utslett, hyppige tilbakefall | Glycyl–tRNA-syntetase | |
| Anti–KS | ~5 %. ILD alene, assosiert med malignitet | Asparaginyl–tRNA-syntetase | |
| Anti–Zo, Ha/YRS, SC, JS, tryptophanyl | <1 %. Klinisk betydning ofte uklar | Sjeldne antistoffer, begrenset dokumentasjon | |
| Myositt-relaterte antistoffer (MRA) | Anti-Ro52 / SSA | Assosiert med økt risiko for ILD ved ASS | Også vanlig ved Sjøgrens og SLE |
| ACPA / anti-CCP | 6–9 %. Assosiert med artritt, særlig tidlig polyartritt | (Gonzalez-Gay MA, 2018; Cavagna L, 2010). | |
| Revmafaktorer (RF) | Forekommer særlig ved leddmanifestasjoner | Nyttig ved differensiering mot RA |

Bildediagnostikk
-CT thoraks/lunger (HRCT) er nesten obligatorisk ved diagnose og i forløpet. Typiske funn:
- Non-spesifikk interstitiell pneumoni (NSIP)– eller organiserende pneumoni (OP)-mønster (78–100 %)
- Mattglassfortetninger
- Etter hvert fibrose eller UIP-liknende (bikake-) mønster (Waseda Y, 2016).
-MR-undersøkelser (oftest) av lårmuskler kan vise muskelødem forenelig med myositt og hjelper å velge biopsisted. Muskelødem er imidlertid ikke spesifikt og kan sees også ved rabdomyolyse og muskeldystrofi. Ved sykdomsdebut kan også fasciitt observeres (differensialdiagnose: eosinofil fasciitt) (Ebbo M, 2013).
-Røntgen av affiserte ledd (oftest fingre). Erosiv artritt ses hos 15-20% av tilfellene. CT- eller MR av aktuelle organer eller PET/CT dersom mistanke om malignitet.
-Røntgen øsofagus med kontrastmiddel under svelgeakt kan vise om dysmotilitet foreligger.

Lungefunksjonstester viser vanligvis et restriktivt mønster med nedsatt total lungekapasitet (TLC) <80% av forventet. Gassdiffusjon (DLCO) og funksjonell vital kapasitet (FVC) som ofte følges opp i klinisk praksis er tilsvarende redusert. Både DLCO og FVC korrelerer vanligvis med utbredelsen av ILD slik en ser den ved HRCT (Andersson H, 2016).
Bronkoskopi med bronkial skyllevæske (lavage / BAL) og transbronkial biopsi er invasive undersøkelser som vanligvis ikke er nødvendig ved ASS. Indikasjon foreligger likevel hvis diagnosen er usikker og særlig ved mistanke om infeksjon (pneumocystis og andre opportunistiske), hypersensitivitetspneumoni eller andre differensialdiagnoser. Ved BAL kan lungelegen vurdere lokale tegn til den inflammatoriske aktiviteten i lungene/bronkiene. I tillegg tas ofte sekret til dyrkning for å utelukke infeksjon (differensialdiagnose).
Elektromyografi (EMG). EMG brukes sjeldnere nå enn tidligere, fordi MR gir mer anatomisk informasjon. EMG kan likevel skille mellom nevrogen og myogen sykdom og styrke mistanken om myositt.
-
EKG og ekkokardiografi ved mistanke om hjertemanifestasjon.
-
Pulmonal hypertensjon forekommer hos ca. 7,9 %. Årsaken er vanligvis lungesykdommen ILD, men pulmonal hypertensjon er likevel en uavhengig risikofaktor for alvorlig sykdomsforløp (Hervier B, 2013).
Muskelbiopsi
Biopsi er ikke alltid nødvendig, men nyttig når diagnosen er usikker. Typiske funn ved ASS:
- Perifascikulær nekrose (ligner dermatomyositt)
- Nekroser i myofibre utenfor perifascikulære områder
- Perimyseal lymfocyttinfiltrasjon (B-celler og CD4+ T-celler)
Endomysiale infiltrasjoner er mindre vanlig og mer typisk for polymyositt og inklusjonslegeme-myositt (Tanboon J, 2023).
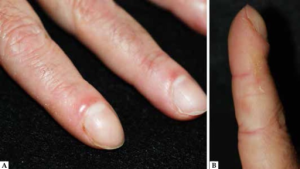
Neglefold-kapillaroskopi. Patologiske funn ses hos >60 %, inkludert:
- Periunguale teleangiektasier.
- Dilaterte eller avbrutte kapillærer.
Vanligst hos pasienter med Raynauds fenomen (Sebastini M, 2019).
6-minutters gangtest. Testen vurderer samlet funksjon av lunger, hjerte, muskler og ledd. Avstanden som tilbakelegges brukes for oppfølging av sykdomsprogresjon. Den forutsetter at pasienten kan gå minst 6 minutter i jevnt, raskt tempo. Resultatet gjenspeiler en kombinasjon av hjerte- og lungekapasitet, ledd og muskelaffeksjon (Enright PL, 2003).
Klassifikasjonskriterier
ASS kan klassifiseres som en egen diagnose ved siden av polymyositt og dermatomyositt eller inkluderes i disse (Marco JL, 2020).
Tabell. Foreslåtte EULAR-ACR klassifikasjonskriterier for antisyntetasesyndrom (ASS) (EULAR, 2025).
|
I hvert klinisk/serologiske domene telles bare den høyeste vektingen. Variabler som ikke er klinisk eller serologisk vurdert skal gis score 0. |
|||
| Kliniske domener | Score | Andre kliniske holdepunkter | Score |
| Leddmanifestasjon | Mekanikerhender/Hikers feet | 1,5 | |
| Inflammatorisk artralgi eller artritt | 1,0 | Raynauds fenomen | 0,5 |
| Muskelaffeksjon | Inflammatorisk utslett (Gottrons, heliotropt, V-tegn, sjal-tegn, sommerfugl-utslett-lignende i ansikt) | 0,5 | |
| Subkliniske tegn på myositt (CK, EMG. MR) | 1,0 | Uforklart feber | 0,5 |
| Kliniske tegn på myositt | 1,5 | Serologisk domene | |
| Biopsi tyder på myositt | 2,0 | Anti-ARS (aminoacyl-tRNA syntetase)- antistoff | |
| Interstitiell lungesykdom (ILD) | Non-Jo-1 positiv* ved non-IP (immunpresipitering)- metoder | 3,0 | |
| UIP eller andre/uklassifiserbart mønster | 1,5 | Jo-1 positiv uansett metode eller non-Jo-1 positiv ved IP | 3,5 |
| Predominant NSIP- og/eller OP-mønster | 2,5 | ANA med cytoplasmatisk mønster | 1,5 |
| anti-Ro52 eller anti-SSA antistoffer | 1,0 | ||
|
Sikkert ASS: Total-score ≥ 5,5 + minst ett klinisk domene + anti-ARS (sens. 94,3%, spes. 99,7%) Sannsynlig ASS: ≥ 5,0 + minst ett klinisk domene; anti-ARS positivitet er ikke obligatorisk (sens 97,5%, spes. 87,6%). Mulig ASS: ≥ 4,0 + minst ett klinisk domene eller et annen klinisk variabel; anti-ARS positivitet er ikke obligatorisk (sens. 98,7, spes. 80,1%). |
|||
*Tilføyelse til tabellen: Non-Jo-1 kan omfatte PL-7, PL-12, EJ og KS ved ELISA-teknikk eller OJ og KS ved Line blot.
Eldre klassifikasjonskriterier
| Sammenligning av to sett foreslåtte kriterier (Marco JL, 2020) | |
| Absolutt krav: Forekomst av anti-tRNA syntetase antistoff |
|
| Connors GR, 2010 |
Solomon J, 2011 |
| Antisyntetase antistoff pluss ett eller flere av følgende: | Antisyntetase antistoff pluss to major kriterier eller ett major- og to minor kriterier Major kriterier: |
| –Myositt ved Bohan og Peter kriterier (Bohan A, Peter JB. Polymyositis, 1975 del I, del II ) | -Myositt ved Bohan og Peter kriterier (se myositt) |
| –ILD uten annen forklaring eller årsak | -ILD uten annen forklaring eller årsak |
| –Artritt -Uforklart, vedvarende feber -Raynauds fenomen | Minor kriterier: |
| -Mekanikerhender (hud) | -Artritt |
| -Raynauds fenomen | |
| -Mekanikerhender | |
Differensialdiagnoser
Lunge-manifestasjoner ved ASS blir ofte misoppfattet som infeksjoner, hypersensitivitets-pneumoni eller idiopatisk lungefibrose, men også andre differensialdiagnoser bør vurderes:
- Infeksjoner: Både virale og bakterielle infeksjoner kan gi feber, hoste og lungeinfiltrater som kan forveksles med lungemanifestasjoner ved ASS.
- Lungefibrose (idiopatisk): Arrdannelse i lungene kan gi pustevansker. Symptomene med tørrhoste og dyspne kan ligne ASS. Diagnosen stilles ved hjelp av CT-bilder og utelukking av andre årsaker.
-
Andre myopatier:
- Polymyositt (PM) og dermatomyositt (DM): Diagnosen stilles ved en kombinasjon av kliniske funn, påvisning av de spesifikke antistoffene og muskelbiopsi.
- Muskeldystrofier og andre nevromuskulære sykdommer: Arvelige muskelsykdommer kan gi progredierende muskelsvakhet. EMG-undersøkelse (elektromyografi) og muskelbiopsi er ofte nødvendig for å skille disse fra myositt (og fra ASS).
- Medikamentelt indusert myopati: Enkelte medisiner som statiner, kan forårsake muskelsmerter og muskelsvakhet.
- Psoriasisartritt: Kan gi artritt i små ledd (inkludert tommel) og hudmanifestasjoner (psoriasis).
- Revmatoid artritt medfører også artralgi, artritt, stivhet og tretthet. ASS kan dessuten være assosiert med RF eller a-CCP antistoff, men RA har typisk symmetrisk erosiv affeksjon av små ledd.
- Sarkoidose. Kan ramme lungene, huden og leddene og gi lungemanifestasjoner og leddsmerter som ligner på ASS.
- Uspesifikke utslag i ASS-antistoff ved polymyositt eller sarkoidose kan også forveksles med ASS.
- Sjøgrens syndrom: Rammer primært eksokrine kjertler (sicca-symptomer), men kan også medføre leddsmerter, tretthet og lungeinvolvering som overlapper med ASS, spesielt ASS når anti-SSA antistoff er tilstede.
- SLE. Leddsmerter, hudutslett og lungeinvolvering som overlapper med ASS, spesielt når anti-SSA antistoff er tilstede.
Svangerskap ved ASS
Kunnskapsgrunnlaget om svangerskap ved ASS er begrenset, fordi tilstanden er sjelden og få tilfeller er rapportert i litteraturen. En systematisk gjennomgang fra 2024 identifiserte kun ti svangerskap hos kvinner med anti-Jo-1-antistoff (Mourot A, 2024). Syv av kvinnene hadde kjent ASS før graviditeten, mens tre fikk diagnosen under svangerskapet. De sistnevnte tilfellene var assosiert med alvorlig matern interstitiell lungesykdom og endte i fosterdød, noe som understreker den betydelige risikoen når ASS debuterer under svangerskapet. Blant de syv kvinnene med etablert diagnose før konsepsjon ble seks svangerskap komplisert av prematur fødsel. Det understreker at gravide med ASS bør følges opp som høyrisikopasienter (Mourot A, 2024).
Dersom anti-SSA (Ro52/SSA) foreligger i tillegg til ASS, anbefales overvåkning etter samme prinsipper som ved Sjøgrens syndrom. Dette inkluderer serielle undersøkelser av føtal hjertefrekvens i uke 16–26, ettersom det foreligger om lag 2 % risiko for kongenitt hjerteblokk, samt vurdering av mulig neonatal lupus etter fødsel (Green LJ, 2020).
Samlet ett antyder tilgjengelige data at risikoen for alvorlige svangerskapskomplikasjoner er særlig høy når ASS diagnostiseres under graviditeten. Begrensede terapeutiske muligheter i svangerskapet kan bidra til dette risikobildet (Mourot A, 2024). Det anbefales at håndteringen av slike svangerskap skjer i tett samarbeid mellom revmatolog, lungespesialist og fødeavdeling med erfaring i høyrisikograviditet. For ytterligere detaljer henvises til nasjonale anbefalinger fra NKSR.
Behandling
Sykdomsforløpet ved ASS er individuelt og behandlingen må tilpasses den enkeltes sykdoms-manifestasjoner (persontilpasset behandling), sykdomsforløp, komorbiditet og toleranse. Likevel er noen råd universelle. Blant non-farmakologiske tiltak er fysikalsk behandling, egentrening og i noen tilfeller er lunge-rehabilitering aktuelt. Før en begynner bør en sette mål som skal nås hvis behandlingen virker etter hensikten (“treat to target”). Målet baseres på reduksjon av sykdomsaktivitet og individuelle manifestasjoner.
Immunsuppresjon er viktig i behandlingen av ASS (med ILD). Behandlingsvalget avhenger av alvorlighetsgrad ved diagnose, sykdomsforløpet, respons på initial terapi, komorbiditet, behandlerens erfaring med tilgjengelige metoder og pasientens preferanser. Valg av medikamenter kan være vanskelig fordi det foreligger relativ få gode studier og ingen medikamenter er spesifikt godkjent for ASS. Behandlingen kan derfor kreve ekstra god pasientinformasjon og oppfølging i henhold til utprøvende behandling.
En kan dele strategien inn i induksjonsbehandling som skal stanse progresjon og redusere inflammasjon, samt en vedlikeholdsbehandling som skal hindre residiv.
Ved mild sykdom med bevarte lungefunksjonstester, minimale symptomer, < 10% involvering på CT og ingen oksygenbehov, kan pasienten overvåkes nøye med lungefunksjonstester hver 3.-4. måned.
Induksjonsbehandling. Kortikosteroider er viktige i de fleste tilfeller, særlig ved ILD. Dosen baseres på sykdommens alvorlighetsgrad. Medikamentet virker raskt , har lav kostnad og god tilgjengelighet. Anbefales ved forverring av symptomer, radiografisk progresjon eller forverring av PFTs (fall på 5% eller 10% for henholdsvis FVC og DLCO).
Tabell. Medikamentell behandling ved antisyntetasesyndrom (ASS)
🟩 Førstelinje / sentrale behandlingstiltak, 🟨 Tilleggs- eller situasjonsavhengig behandling, 🟥 Behandling ved refraktær sykdom / utprøvende behandling.
| Tiltak | Behandling | Dose / bruk | Kommentarer / indikasjoner | Nivå/fargekode |
|---|---|---|---|---|
| Kortikosteroider | Prednison; metylprednisolon | Prednison 0,75–1 mg/kg/dag. Ved akutt forverring: Metylprednisolon 500–1000 mg ×3 dager → prednison 1 mg/kg/dag (maks 60 mg). | Førstevalg ved ASS-ILD. Brukes ved symptomforverring, radiologisk progresjon eller PFT-fall. | 🟩 (Mimori T, 2012); Hallowell RW, 2023) |
| CellCept | 2000–3000 mg/dag i to doser | Foretrukket steroid-sparende behandling ved ILD pga. best toleranse. | 🟩 (Huapaya JA, 2019) | |
| Imurel | 2–2,5 mg/kg/dag | Effektivt steroid-sparende alternativ. | 🟩 (Huapaya JA, 2019) | |
| Metotreksat (MTX) | MTX | 15–25 mg/uke | Effektivt ved myositt og artritt. Brukes med forsiktighet ved ILD pga. pneumonitt-risiko. | 🟩 (McGrath ER, 2018) |
| Cyklofosfamid | Sendoxan | Individuelt regime | Brukes ved alvorlig eller raskt progredierende ILD. | 🟨 (Huapaya JA, 2019) |
| Calcineurinhemmere | Takrolimus (foretrukket), ciclosporin) | Takrolimus 0,5–1 mg ×2 | Brukes ved behandlingsrefraktær ILD. Takrolimus har bedre effekt/bivirkning-profil. | 🟨 (Labirua-Iturburu A, 2013; Witt LJ, 2016). |
| IVIG; Immunglobulin. | Intravenøs immunglobulin | 2 g/kg/mnd fordelt over 3–5 dager | Tillegg ved refraktær ILD eller myositt. Obs: trombose, væskeoverbelastning, aseptisk meningitt. | 🟨 (Oddis CV, 2018) |
| Antifibrotisk behandling | Nintedanib, pirfenidon | Standard doser | Vurderes ved UIP-mønster. Immunsuppresjon er fortsatt primær behandling. | 🟨 (Hallowell RW, 2023) |
| Rituksimab | MabThera | 1000 mg dag 0 og 14; gjentas hver 6. mnd | Effektivt ved ASS-ILD. Best respons ved kort sykdomsvarighet, eksaserbasjoner og anti-SSA. Infeksjonsrisiko! | 🟥 (Sem M, 2009; Bauhammer J, 2016) |
| JAK-hemmere. | Tofacitinib | Ikke etablert standarddose | Lovende ved myositt og MDA5-ILD, men begrenset data og kardiovaskulær risiko. Ikke rutinebehandling. | 🟥 (Hallowell RW, 2023) |
| Lungetransplantasjon | — | — | For alvorlig, behandlingsrefraktær, progredierende ILD. | 🟥 (Mimori T, 2012) |
Vedlikeholdsbehandling
-csDMARDs. I milde eller moderate tilfeller med myositt velges ofte metotreksat (15-20mg/uke), azathioprin (150mg/d) eller mykofenolat 100-1500 mg to ganger i døgnet) (McGrath ER, 2018). Metotreksat unngås imidlertid ofte ved ILD siden lungebivirkning forekommer (pneumonitt, sjelden) og da kan være vanskelig å skille fra sykdomsprogresjon.
–Biologisk. Rituksimab. Ved alvorligere sykdomsforløp kan mer aggressiv vedlikeholdsbehandling være nødvendig med calcineurin-hemmere eller rituksimab, sjelden cyklofosfamid.
–Immunglobulin. Det er publisert et økende antall kasusrapporter og serier som antyder nytten av IVIG som tilleggsterapi hos pasienter med ILD, spesielt ved refraktær sykdom. Doseringen er ofte 2 g/kg/måned fordelt over 3-5 dager (Oddis CV, 2018). Venøs tromboemboli, væskeoverbelastning, hodepine (aseptisk meningitt), antistoffmedierte cytopenier, anafylaksi, infusjonsreaksjoner kan være bivirkninger.
Oppfølging Behandlingen bør ha spesiell fokus på å redusere lunge-infiltrater ved NSIP type. Også undersøkelser i forløpet er viktig fordi 67% som initialt var uten ILD kan utvikle lunge-manifestasjonen i forløpet (Cavagna L, 2015). I oppfølgingen inngår ofte:
- Lungefunksjons-tester
- 6-minutters gangtest
- HRCT av lunger
- Ved mistanke om pulmonal hypertensjon er ekkokardiografi aktuelt.
- Muskelstyrken kan vurderes med standardiserte målinger i regi av fysioterapeut.
Prognose
Prognosen ved ASS varierer betydelig og avhenger i stor grad av graden av lungemanifestasjoner, typen myosittspesifikt antistoff og hvor tidlig sykdommen blir diagnostisert og behandlet. ASS regnes som en potensielt alvorlig sykdom, men med tidlig identifikasjon og adekvat immunsuppressiv behandling kan mange pasienter oppnå god symptomkontroll og bevare funksjon over tid. Prognosen er generelt best hos pasienter som ikke utvikler alvorlig ILD.
Betydningen av lungemanifestasjoner. ILD er den viktigste determinanten for langtidsutfall ved ASS. Progressiv fibrosedannelse kan føre til betydelig redusert lungefunksjon og er den hyppigste dødsårsaken i flere kohorter. Kliniske tegn på forverring, som økende dyspné og hoste, må vurderes nøye, særlig fordi pasientene samtidig har økt risiko for alvorlige luftveisinfeksjoner. Denne infeksjonsrisikoen forsterkes av svelgevansker og aspirasjonstendens, samt av progredierende svakhet i respiratorisk muskulatur, som også kan bidra til dyspné.
Overlevelse og prognostiske faktorer. I en kohortstudie av 202 pasienter med ASS publisert av Aggarwal et al. (2014), ble det rapportert at pasienter med anti-Jo-1-antistoff hadde en 5-års overlevelse på 90 % og en 10-års overlevelse på 70 %. Pasienter uten anti-Jo-1 hadde dårligere prognose, med 5- og 10-års overlevelse på henholdsvis 75 % og 47 %. Lungefibrose var den vanligste dødsårsaken i begge grupper (Aggarwal R, 2014).
Betydningen av antistoffprofil. Antistoffprofilen har diagnostisk og prognostisk betydning. Pasienter med anti-Jo-1-antistoff har generelt et bedre langtidsforløp og bedre respons på behandlingen sammenlignet med pasienter med andre anti-aminoacyl-tRNA-syntetase-antistoffer (anti-ARS). Spesielt anti-PL-7 og anti-PL-12 er assosiert med mer alvorlig og raskt progredierende ILD, og dermed dårligere overlevelse.
Retningslinjer, anbefalinger, prosedyrer
Litteratur
- Faghihi‐Kashani S, 2025
- Patel P, 2024
- Hallowell RW, 2023
- Wells M, 2022
- Alfraji N, 2021
- Marco JL, 2020
- Andersson H, 2016
- Trallero-Araguas E, 2016
- Witt LJ, 2016 (behandling)
- Bauhammer J, 2016 (behandling med rituksimab)
Teksten er skrevet og gjennomgått av forfatterne. I bearbeidelsen er kunstig intelligens brukt i noen avsnitt.
