VASKULITT (REV 034-052)
89 Granulomatose med polyangiit (GPA) tidligere Wegeners granulomatose (REV 034)
Ragnar Gunnarsson
Kjennetegn på GPA
Symptomer fra øvre luftveier (bihuler, nese, trachea) med blodige skorper fra nese og/eller redusert hørsel/otitt ved affeksjon av mellomøre/mastoid celler.
Affeksjon av lunger forekommer often uten særlige symptomer (Husk: CT thorax).
Nyreaffeksjon med pauci immune glomerulonefritt som ubehandlet medfører redusert nyrefunksjon og nyresvikt (Husk: urin-undersøkelse med mikroskopisk hematuri og/eller nefritisk urin sediment).
Øyesymptomer som oftest med episkleritt, hudvaskulitt og nerveaffeksjon oftest med mononeuritis multiplex er de øvrige de mest vanlige symptomene.
Almenn symptomer som feber, vekttap og utmattelse. Leddsmerter og leddbetennelse.
Høye inflammasjonsparametere (SR og CRP) samt påvist anti-PR3-ANCA IgG i blodprøve hos de aller fleste med aktiv sykdom
Diagnosekoder ICD-10: (Tiende versjon av den internasjonale statistiske klassifikasjonen av sykdommer og beslektede helseproblemer i regi av WHO (ICD10) M31.3 Granulomatose med polyangiit (GPA)
Prosedyrekoder: 6-minutter gangtest: FYFX05. Intravenøs infusjon: WBGM00. Infusjon med gammaglobulin: RPGM05. Intravenøs infusjon med cytostatika: WBOC05. EKG: FPFE15
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A. Behandling med rituksimab: L01XC02. Behandling med cyklofosfamid: L01AA01.
Definisjon
Granulomatose med polyangiit (GPA) karakterisert av nekrotiserende granulomatøs vaskulitt av små og mellomstore kar. Sykdommen rammer i hovedsak øvre og/elle nedre luftveier, men kan ellers ramme kar i de fleste andre organsystemer i tillegg som oftest i nyrer og hud.
GPA er sterkt assosiert til antistoff mot granuler i cytoplasma til neutrofile granulocytter. Et autoantistoff, anti-neutrofil cytoplasma antistoff (ANCA), bindes spesifikk på proteinase 3 (PR3-ANCA) og er sterkt assosiert til GPA. Histologisk kan granulomer sees.
Genetikk og patogenese
Granulomatose med polyangiit (GPA), mikroskopisk polyangiit (MPA) samt og eosinofil granulomatøs polyangiit (EGPA) er klassifiserte som ANCA assosierte vaskulitter (AAV). Årsaken til at den enkelte pasienten utvikler GPA er ukjent. Det er data som knytter dette til bærerstatus av bakterien Staphylococcus aureus. Det har vært funnet assosiasjon til medikamenter som; propylthiouracil og sulfasalazin ved AAV like tilstander. I tillegg ved bruk av kokain gi affeksjon av nese og bihuler som kalles «cocain indusert midline destrucitve lesions» forkortet til CIMDL. Pasienter med CIMDL kan ha positiv PR3 ANCA, men ulikt GPA har flertall i tillegg også ANCA rettet mot human elastase (HNE-ANCA) og anti-MPO ANCA.
Ved utvikling av granulomatose fester nøytrofile leukocytter seg til endotel utskiller de oksygenrike radikaler og proteolytiske enzymer (inkludert proteinase 3/PR3) som er toksiske for endotelcellene. I tillegg utløser nøytrofile leukocytter ekstracellulære nett (NETs) med PR3. Frigjøring og binding av antistoffer mot PR3 (PR3-ANCA), fører til aktivering av den alternative komplement aktiveringsveien. Det leder til forsterkning av lokal inflammatorisk respons og øker vevsskade.
Granulom dannelse er et av de histologiske trekkene ved GPA, men sees ikke ved MPA. Granulomer antas å være relatert til cytokinsekresjonsmønster av type 1 T-hjelpe celler (Th1). Den dominerende reaksjonen ved GPA er via Th2-type reaksjon som via IL-4 og IL-10 fører til «vaskulittiske» systemiske manifestasjoner som for eksempel alveolare blødninger og glomerulonefritt. Andre typer lymfocytter og cytokiner er også involvert i denne inflammasjonskaskaden som Th17-lymfocytter.
")
MPA og GPA har forskjellig genetisk assosiasjoner som tyder på at dette er separate sykdommer som deler felles kliniske og patologiske trekk. GWA-studier («Genome-Wide Association Studies») bekreftet forskjell mellom GPA og MPA som ikke er assosiert til den kliniske fenotypen, men assosiert til antistoff tilknytningen, dvs. om de er anti-PR3-ANCA eller -MPO-ANCA positive. HLA-DPB1*0401, PRTN3 (Proteinase 3 Neutrophil Serine Protease), SERPINA1 (Alfa1 antitrypsin: serine protease inhibitor) er assosiert til PR3-ANCA (1-4).
Et betydelig problem ved forsking på patogenese av PR3-ANCA vaskulitt er at man foreløpig ikke har bra dyremodell, som man har ved anti-myeloperoksidase ANCA (MPO-ANCA) vaskulitt. Der anti-MPO-ANCA kan indusere vaskulitt i relativt enkle dyremodeller.
Epidemiologi
Forekomst (insidens) av GPA er estimert 2–12 tilfeller per år per million innbyggere og utbredelse (prevalens) 23–160 tilfeller per million innbyggere. GPA påvirker begge kjønn relativt likt. Median alder for pasienter ved diagnose er i det femte tiåret. Barn kan i svært sjeldne tilfeller også bli rammet. I Europa ser forekomsten av GPA ut til å vise en nord-sør-gradient. Rapporterte forekomsten av GPA dobbelt så høy i Norge som i Spania (10,6 mot 4,9 nye tilfeller per år per million innbyggere). En interimsanalyse av «Diagnostic and Classification Criteria in Vasculitis Study» DCVAS AAV-pasientdata indikerer at PR3-ANCA AAV er den dominerende typen av vaskulitt hos nordeuropeere, samt innbyggere i Midtøsten og i det indiske subkontinent, mens MPO-ANCA AAV er den dominerende hos øst asiatiske, dvs. kinesisk, koreansk og japanske (5).
Kliniske forhold
. CC BY 2.0")
De tre viktigste målorganene ved GPA er øvre (ØNH) og nedre (lunger) luftveier og nyrer. Det er to epidemiologiske studer på GPA, en fra Frankrike og den andre fra Tyskland med 396 og 445 pasienter, publisert i 2010 og 2013. Fordelingen av de kliniske manifestasjonene ved systemisk sykdom angitt nedenfor kommer fra disse to studiene (6, 7).
Sykdomsforløpet ved GPA kan variere. Noen pasienter kan i flere år ulmende plager med residiverende nesetetthet, sinusitt og otitt. Det er anført at 5% av pasientene har et slikt vedvarende tidligere kaldt begrenset sykdomsbilde (8).
Allmennsymptomer. Fleste pasienter har allmennsymptomer og beskriver nedsatt almenntilstand og det kan følge vekttap og utmattelse og noen har lavgradig feber. Leddsmerter er hyppige, og det er rapporter om nondestruktiv artritt i debut fase av sykdommen, men de symptomene responderer som oftest raskt på behandling.
ØNH symptomer (85-95%): Epistaxis (neseblødning), nasale ulcerasjoner og skader i form av neseseptumperforasjon og sadelnesedeformitet. Sinusitt med beinnydannelse. Affeksjon av munnslimhinne med gingival hyperplasi kan forekomme i sjeldne tilfeller. Otologiske symptomer: Otitis media, hørselstap, kondritt, otitis eksterna og granulomer i membrana tympanica samt betennelse i ørebensknuten og mastoidcellene (mastoiditt).
Lungeaffeksjon (50-77%): Nekrotiserende granulomatøs betennelse i lunger har karakteristiske patognomonisk histopatologi som inkluderer; neutrofil mikroabscesser, fibrinoid nekrose, palisade-dannende histiocytter med kjempeceller, som danner et granulomatøst betennelsesmønster som ofte kalles “geografisk nekrose”. Radiologisk korrelerer den nekrotiserende granulomatøse betennelsen med en eller flere lungeknuter med eller uten kavitasjon og lungeinfiltrater. Hoveddifferensial diagnosene ved lungeforandringene ved GPA er bla. primær lungecancer eller metastaser, lymfoproliferative sykdommer inkl. lymfomatoid granulomatose, som er et sjeldent angiosentrisk og angiodestruktiv Epstein Barr-virusrelatert lymfom. Andre differensialdiagnoser er bla. lunge sarkoidose og lungeaffeksjon av autoimmune sykdommer. Infeksjoner inklusivt soppinfeksjoner er en viktig differensial diagnose (histoplasmose, coccidiodomycosis ofl.).
Granulomer i lunger trenger ikke å gi særlige kliniske symptomer. Ved mistanke om AAV bør man ha en lav terskel å utføre computer tomografi (CT thoraks) av lunger. Affeksjon av luftveier med affeksjon av trachea og bronkier gir etter hvert kliniske manifestasjoner i form av dyspne, stridor, hoste og hemoptyse. Alveoler blødninger er en hoveddødsårsak av sykdommen.
Diffus alveolar hemoragi (DAH) som kan oppstå i opp mot 25% av pasienter med GPA og MPA. Tilstanden kan bekreftes med å påvise blod ved bronkoskopi med skylling (bronchoalveolar lavage, BAL) også for å utelukke anti-GBM antistoff/Goodpasture syndrome, samt og infeksjon som for eksempel Pneumocystis jiroveci pneumoni (PJP).
Interstitial lungesykdom (ILD). MPO-ANCA positive AAV pasienter er i på den andre siden i økt risiko for å utvikle interstitial lungesykdom med lungefibrose, det som kalles «usual interstitial pneumonia» (UIP). UIP utvikling ved er assosiert til økt alder (>65 år), tidligere alveolar blødning, men ikke valg av immunsuppresjon og er en selvstendig negativ risikofaktor for prognose og vises til en nylig case-control studie fra Frankrike (9).
Tracheobronkial betennelse er assosiert til GPA. Tilstanden er utfordrende. Bronkoskopi den mest viktige undersøkelsesmetoden for å kartlegge tilstanden og ta dyrkninger og histologi. Tracheobronkial betennelse er ofte terapiresistent ved systemisk immunsuppresjon og tilstanden gjenkjennes ofte lavgradig vedvarende betennelse med arrdannelse og stenosering i luftveier. Det kan forekomme skade på brusk i luftveier og luftveiskollaps med trakeal- og/eller bronkomalasi og i tillegg kan dette være komplisert med sekundær infeksjoner. Differensialdiagnosen ved tracheobronkial betennelse er bl.a. relapsing polykondritt.
")
Nyreaffeksjon (50-77%): mikroskopisk hematuri med nefrittisk urinsediment oftest granulære og erytrocyturi med fallende nyrefunksjon og utvikling av nyresvikt. Nyrebiopsi viser rask progressiv glomerulonefritt (RPGN) av type III. (Type I RPGN sees ved anti-GBM sykdom (Goodpasture) og type II RPGN sees ved immunkompleks medierte tilstander.) Det sees nekrotiserende ekstrakapillær fibrocellulære halvmåner med pauciimmun glomerulonefritt. Avhengig av utvikling og tidspunkt for biopsi vil det påvises permanent skadetegn med sklerotiske glomeruli og interstitial fibrose i nyrevev. Nyrebiopsi kan både bekrefte diagnosen og har en avgjørende betydning i å forutse prognose og behandlingsvalg. Ved AAV kan patologien deles ned i fire klasser (10), i fokal affeksjon (≥50% av glomeruli uten patologi), halvmåneformet («crescendic») affeksjon (≥50% av glomeruli med cellulære halvmåner), blandet (mixed) glomerulær affeksjon (<50% normal, <50% halvmåner, <50% sklerotiske) og sklerotisk glomerulær affeksjon (≥50% global skleroserte). Prognosen er best hos de som klassifiseres med fokal glomerulære forandringer og verst hos de som klassifiseres med sklerotiske glomerulære forandringer som er i grunn permanent skade.
Hudaffeksjon (20-30%): ved AAV sees i form av vaskulær purpura, livedo reticularis, pustler, blemmeformete lesjoner med vesikler eller noduli, ulcerøse og/eller nekrotiske sår og subkutane knuter. Histologi viser som regel leukocytoklastisk vaskulitt og ofte sees fibrinoid nekrose i karvegg. Hudbiopsier ved GPA er svært sjelden diagnostiske for GPA (eller MPA) og viser svært sjelden karakteristisk granulomatøs granulomatøs inflammasjon.
Perifer nevropati (23-28%): forårsakes av iskemisk aksonal vaskulitt av vasa nervorum. Vanligst er mononevritis multipleks mens sensomotorisk polyneuropati er mindre vanlig.
")
Central nervøse manifestasjoner av GPA er svært sjeldne og som oftest følge av invasjon fra bihuler eller mastoid celler. Det svært sjelden med vaskulitt av intracerebrale kar. Det er hyppigere med affeksjon av dura matter med pachymeningitt med som oftest affeksjon av hjernenerver.
Øyeaffeksjon (35-40%): i form av konjunktivitt, episkleritt, uveitt, retrobulbært granulom og optikusnevritt.
Annet: Sjeldne manifestasjoner er; koronar angiitt, pankarditt og iskemi av tynn- og/eller tykktarm. Det er betydelig økt fare for venøse tromboembolier (VTE) med både dypvenetrombose og lungeemboli ved aktiv AAV (11).
Laboratoriefunn
Høy senkningsreaksjon (SR) og forhøyet C-reaktiv protein (CPR) med normocytær normokrom anemi og trombocytose som gjenspeiler forhøyet inflammasjonsaktivitet sees hos aller fleste som tegn på generalisert inflammasjonsaktivitet. Det er viktig å ta utføre differensial telling av leukocytter med tanke på eosinofili og andre differensialdiagnose. Procalcitonin er vanligvis normal ved AAV men kan være moderat forhøyet.
Det er svært viktig å ta nyrefunksjonsparametere og forsikre seg urinprøve for urinstiks og mikroskopi.
ANCA screening med testing for PR3- og MPO- ANCA og på OUS går det an å få ANCA test i løpet av få timer hvis kliniske situasjonen krever det og da evt. også anti-GBM hvis kliniske bildet er forenlig med Goodpastures syndrom som er en immunologisk betinget sykdom som skyldes auto-antistoff mot basalmembranen i lunge og nyrevev.
Diagnose
. CC BY-NC 4.0")
Diagnosen av GPA stilles på mønstergjenkjenning. Der det kliniske sykdomsbilde støttes opp av kliniske funn og samt blodprøver inklusiv ANCA og eventuelt urin og aktuelle radiologisk funn (CT/MRI bihuler, tinningben og CT lunger/luftveier). Det foretrekkes at diagnosen blir histologisk verifisert med biopsi fra som oftest neseslimhinne, lunge/luftveier og eller nyre. Histologisk påvises vaskulitt, granulomer, granulomatøs inflammasjon og/eller pauciimmun glomerulonefritt. Det er viktig at neseslimhinnebiopsi tas dypt ned til brusk, der det er relativ stort antall av neseslimhinnebiopsier er ikke representative.
Differensial diagnoser ved GPA
- Annen ANCA vaskulitt som EGPA eller MPA. I tillegg Goodpasture/anti-GBM syndrom.
- Medikament utløste tilstander, for eksempel. propyltiouracil (12) utløst tilstand. Ulovlige midler inkludert kokain og levamisol som er ofte brukt som tilsetningsstoff til kokain kan gi AAV liknende tilstand. Det er svært viktig at man for pålitelig medikament evt. rusmiddel anamnese og evt. urin/blodprøve, hvis nødvendig.
- Infeksjoner (bakterier og sopp). Inklusiv bakteriell endokarditt.
- Lymfom.
- Sarkoidose.
- IgG4 assosiert sykdom (IgG4-RD).
- Graves sykdom.
- Orbital pseudotumor.
- Relapsing polykondritt kan utløse destruksjon med utvikling av sadelnese og/eller stenoseutvikling i luftveier men GPA gir ikke bihule ytre øreaffeksjon.
- Kolesterolembolier, antifosfolipid antistoff syndrom, atrialt myksom kan gi vaskulitt liknende tilstand.
- Letalt midtlinjegranulom / nasalt NK-celle lymfom.
Klassifikasjon, estimering av sykdomsaktivitet og skade
Betegnelsen “begrenset” (limited) GPA ble introdusert i 1966 og ble opprinnelig definert som GPA som ikke involverte nyrene. Denne definisjonen bør unngås fordi begrepet ofte blir tolket feil. “Begrenset” GPA betyr ikke det samme som “ikke-alvorlig” GPA. De som oppfyller definisjonen av “begrenset” GPA kan ha alvorlig livstruende sykdom, og de som ikke oppfyller definisjonen kan ha ganske mild sykdomstilstand.
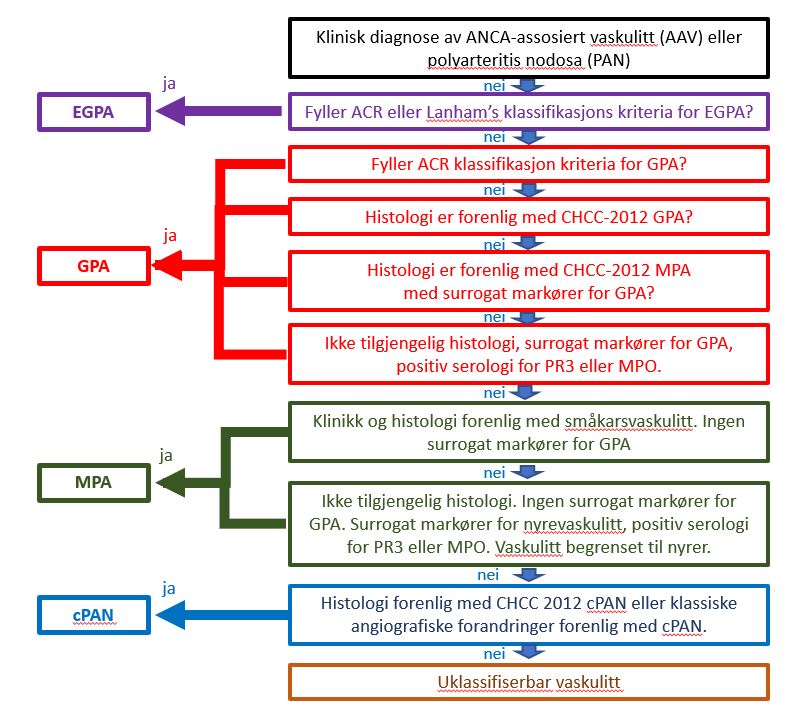
Tidligere klassifikasjonskriterie for GPA (tidligere Wegeners granulomatose) er fra 1990 har vært brukt i flere over 30 år. Det er viktig å være klar over at ANCA status var der ikke inkludert (13). Det forelå på det tidspunkt ikke klassifikasjonskriterie for mikroskopisk polyangiitt (MPA) men det var tidligere vært klassifisert som polyarteritis nodosa (PAN). ANCA assosierte vaskulitter (AAV) var fra 2007 segregert til EGPA, GPA, MPA og polyarteritis nodosa (PAN) og uklassifisert vaskulitt ved hjelp av flytskjema algoritme anbefalt bl.a. av European Medicines Agency (EMA) (Figur 1) (14). Dette er nå erstattet med særskilte klassifikasjonskriteria for GPA, MPA og EGPA med de nye oppdaterte 2022 ACR/EULAR klassifikasjonskriteria for ANCA assosierte vaskulitter (15-17).
Tabel 1: “American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. “
- Betennelse i nese og/eller munn. Utvikling av smertefulle eller smertefrie sår i munn eller purulent eller skorper i nese.
- Lungeforandringer. Radiologiske bilder som viser knuter, infiltrater og/eller kavitering.
- Mikroskopisk hematuri. Mikrohematuri (> 5 røde blodlegemer per synsfelt) eller røde blodlegemesylindrer.
- Granulomatøs betennelse I vevsbiopsi. Histologiske endringer som viser granulomatøs betennelse i arterievegg eller i det perivaskulært og/eller ekstravaskulært.
Tilstanden ble klassifisert som Wegeners granulomatose (=GPA) hvis minst 2 av disse 4 kriteriene er til stede. Tilstedeværelsen av et hvilket som helst to eller flere kriterier gir sensitivitet på 88,2% og spesifisitet på 92,0%. I følge ACR-kriteriene skilles det ikke mellom GPA (Wegeners granulomatose) og MPA, mens derimot Chappel Hill konsensuskriteriene definerer de to som separate sykdommer.
Figur 1. EMA algoritme ved klassifikasjon av ANCA-assosiert vaskulitt og polyarteritis nodosa som tidligere ble brukt (14).
ACR: American College of Rheumatology; CHCC-2012 : Chapel Hill Consensus Conference 2012; cPAN : classic polyarteritis nodosa; EGPA : eosinofil granulomatose med polyangiitt; GPA : granulomatose med polyangiitt; MPA : mikroskopisk polyangiitt. MPO : myeloperoksiadase; PR3 : proteinase 3.
Figur 2. ACR/EULAR klassifikasjonskriteria for granulomatose med polyangiit (GPA) (15).

Sykdomsaktivitet ved GPA som ved MPA og EGPA vurderes etter Birmingham Vasculitis Activity Score versjon 3. Der er maksimal score på 63 poeng. Ved vedvarende sykdom er det maks. score på 33 poeng. Komplett remisjon angis som BVAS =0 med per oral Prednisolon 7,5 mg eller lavere mens vedvarende «sustained» remisjon som BVAS = 0 i 6 måneder eller mer. Kalkulator for BVAS v3 og på PDF-fil. (18, 19).
Skadeomfang vurderes etter Vasculitis Damage Index (VDI) (20). Skåring i VDI er permanent og poeng beholdes, men skaden må ha stått i minimum 3 måneder. Kalkulator VDI.
Behandling
Det vises til de franske anbefalingene som ble publisert i 2020 fra den fra den franske vaskulitt foreningen GFEV, Groupe Français D’étude des Vascularites (21), samt nyeste oppdaterte retningslinjer fra American College of Rheumatology (ACR) og Vasculitis Foundation (VF)som ble publisert i august 2021, 2021 ACR/VF Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis (22) De nyeste Europeiske retningslinjene fra European League Against Rheumatism (EULAR). EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update, som ble publisert tidlig i 2024 (23) og er etter mitt syn de aller beste retningslinjene som er tilgjengelig i dag. Det var også tidlig i 2024 publiserte fra nefrologisk ståsted,Kidney Disease: Improving Global Outcomes (KDIGO) 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis (48).
Viser nærmere til de norske behandlingsretningslinjer fra Norsk Revmatologisk forening (NRF) fra 2024.
I. INDUKSJONSBEHANDLING
A: Ved alvorlig systemisk ny-diagnostisert eller alvorlig residiv av GPA
Første valg ved induksjonsbehandling ved GPA:
Generelt innledes behandlingen med høydose steroid behandling. Som oftest intravenøs metylprednisolon/Solu-Medrol® 500-1000 mg daglig i 3 dager. Etterfulgt av per oral Prednisolon behandling etter PEXIVAS protokollen ((24) (tabell 2). Dette har også vært tatt opp i de norske retningslinjene i behandling av ANCA assosierte vaskulitter og støttes også i siste ACR og EULAR retningslinjene (22, 23). Vært er å få med seg av den såkalte «lavdose» PEXIVAS steroid behandling har kun vært testet hos pasienter som har fått enten cyklofosfamid eller rituksimab (RTX) som induksjonsbehandling, men ikke ved annen induksjonsbehandling som; mykofenolat (MMF) eller metotreksat (MTX). Det bør også nevnes at det var det betydelig overvekt av pasientene i PEXIVAS studien som fikk cyklofosfamid enten intravenøst eller per oralt og kun litt over 15% fikk RTX som induksjonsbehandling (24) og resultatet er foreløpig ikke bekreftet i andre studier.
Tabell 2. Prednisolon nedtrapping etter lavdose PEXIVAS protokoll (24).
| Uke | <50kg | 50-75kg | >75kg |
| IV SoluMedrol 500-1000mg i 3 dager | |||
| 1-2 | 25 | 30 | 40 |
| 3-4 | 20 | 25 | 30 |
| 5-6 | 15 | 20 | 25 |
| 7-8 | 12,5 | 15 | 20 |
| 9-10 | 10 | 12,5 | 15 |
| 11-12 | 7,5 | 10 | 12,5 |
| 13-14 | 6,25 | 7,5 | 10 |
| 15-16 | 5 | 5 | 7,5 |
| 17-18 | 5 | 5 | 7,5 |
| 19-20 | 5 | 5 | 5 |
| 21-22 | 5 | 5 | 5 |
| 23-52 | 5 | 5 | 5 |
| >52 | Individuell behandling |
Avakopan (Tavenos®): er molekyl som gis i tablettform og binder seg opp til komplement C5a reseptor 1 (C5aR1) og hindrer komplementfaktor C5a å sette seg på og stimulere reseptoren. ADVOCATE studien inkluderte 331 pasienter med MPA og GPA, og den er inntil nå verdens nest største behandlingsstudie på AAV der den kommer etter PEXIVAS studien med 704 pasienter inkluderte (34). Hemming av deler av komplementsystemet ser ut til å være paradigmeskifte i behandling av GPA og MPA og kan erstatte glukokortikoid behandlingen, som i stor grad forårsaker økt antall infeksjoner og komplikasjoner hos pasienter med AAV. Pasientene i ADVOCATE studien ble etter induksjonsbehandling randomiserte til å få enten konvensjonell per oral prednisolon behandling som ble faset ut i etter uke 21 og ble sammenlignet med per oral avacopan 30 mg x2 i kombinasjon med standard induksjonsbehandling med rituksimab eller cyklofosfamid. De primære endepunkter, remisjon i uke 26, viste ingen statistisk forskjell mellom avakopan (72,3%) og prednison (70,1%), men i uke 52 var signifikant bedre effekt av avakopan (65,7%) sammenlignet med prednison (54,9%). Risikoen for tilbakefall var signifikant større i gruppen som fikk per oral prednison. I tillegg viste studien det oppsiktsvekkende funn, at pasienter med AAV og nyreaffeksjon ved baseline som ble randomisert til avacopan hadde en signifikant større økning av eGFR sammenlignet med de som fikk prednison, der forskjellen var størst hos pasienter med en baseline eGFR < 20 ml/min /1,73 m2 (35) i tillegg har det vært påvist effekt på AVV pasienter som var i dialyse (36), men disse var ekskludert ADVOATE studien (34).
Avacopan var registrert av FDA for bruk i USA fra oktober 2021 og fra 2022 i Norge under navnet Tavenos®. Avakopan behandling er inkludert i de nyeste Europeiske behandlingsretningslinjene ved behandling av GPA og MPA der de kan erstatte per oral glukokortikoid behandlingen (23). Foreløpig er avakopan ikke uten videre tilgjengelig til norske pasienter hverken på H-resept eller §2. Beslutningsforum for nye metoder konkluderte den 28.8.2023 at det ikke var dokumentert at klinisk nytte stod i rimelig forhold til pris sammenlignet med per oral prednisolon behandling som er som kjent svært billig. Avakopan må søkes på individuell basis til medisinskfagdirektører særskilte grupper som vil profitere på avakopan/Tavenos og glukokortikoidfri behandling. Dette inkluderer bl.a. pasienter med; nyresvikt, diabetes mellitus, overvekt, osteoporose og pasienter med særlig økt infeksjonsrisiko som svært gamle og skrøpelige pasienter. Der avakopan vil sannsynligvis kunne redusere alvorlige komplikasjoner og forhindre langvarige innleggelser.
Plasmaferese ved GPA. Tidligere har det vært vanlig å gi pasienter med alvorlig nyreaffeksjon og/eller lungeblødninger som følge av AAV plasmaferese som er fortsatt standard behandling ved Goodpasture/anti-GBM syndrom. PEXIVAS us, er den største ANCA vaskulitt studien som har vært utført med inkluderte 704 pasienter, viste ingen fordel av plasmaferese ved lungeblødninger hos pasienter med GPA og MPA (24), eller signifikant effekt av plasmaferese ved nyreaffeksjon. Det er antydning til at plasmaferese kan minske risikoen for nyresvikt hos de som har kreatinin over 300 i metaanalyse som innebærer PEXIVAS samt flere mindre og eldre studier som anvender cyklofosfamid, men på den andre siden øker plasmaferese risikoen for alvorlig infeksjon (23, 24, 25).
Rituksimab (RTX): Anti-CD20 antistoff behandling med intravenøst rituksimab behandling er per dags dato første valg ved anti-PR3 antistoff positiv GPA. Rituksimab er kimært mus/humant monoklonalt antistoff, som består av et glykosylert immunglobulin human IgG1 konstant region og variable regioner fra mus. I to randomiserte studier; RAVE og RITUXIVAS ble den såkalte «lymfomprotokoll» brukt. Da gis rituksimab intravenøs ukentlig til sammen i fire uker i doseringen 375mg/m2 overflate (26, 27). Fleste bruker den såkalte reumatoid artritt protokoll som består av intravenøs rituksimab 1.000 mg gitt 2 ganger med 2 ukers intervall. Det har ikke vært utført sammenligningsstudie på disse to protokollene (28) når det gjelder induksjonsbehandling.
Vanlig prosedyre ved behandling med anti-CD20 antistoff behandling som rituksimab er å gi premedikasjon 30-60 min før rituksimab behandling. Den gjenstår i per oral paracetamol 1.000 mg x1, i tillegg histamin H1-reseptorantagonist, enten per oral cetirizin (10mg x1), eller intravenøs deksklorfeniramin 5 mg/ml 1ml IV. I tillegg gis oftest intravenøst metylprednisolon/Solu-Medrol® 125 mg.
Det er dokumentasjon for bedre effekt av rituksimab (RTX) en cyklofosfamid/azathioprin (CYC/AXZA) hos PR3-ANCA positive pasienter i RAVE studien (26). komplett remisjon etter 6 måneder sammenlignet hos de som fikk RTX versus CYC/AZA var på 65% vs. 48% (p=0,04) med odds ratio på 2,11 (95% CI 1,04 – 4.30) (29). Hos pasienter som har ikke kommer i remisjon eller får residiv etter induksjonsbehandling med cyklofosfamid er rituksimab klart første valg. Generelt er det et første valg hos pasienter i reproduktiv alder i tillegg hos de som tidligere har hatt malign sykdom.
B: Annet valg ved induksjonsbehandling ved GPA
Cyclofosfamid (Sendoxan®): er et alkylerende cytostatikum. Medikamentet er en sennepsgassanalog om griper inn i G1- og S-fasen i cellesyklusen med å alkylere DNA-kjedene og hemme immunologisk respons. Intravenøs eller per oral cyklofosfamid evt. er et godt kjent behandlingsalternativ for AAV.
-Intravenøs cyklofosfamid behandling:
Fleste bruker intravenøs cyklofosfamid etter CYCLOPS/EUVAS protokollen (30). Det er generell trend til å bruke intravenøs cyklofosfamid der dette gir lavere kumulativ dosering en ved per oral behandling. Standarddoseringen ved intravenøs behandling er 15 mg/kg med maksimal dosering på 1.200 mg cyklofosfamid per behandlingsrunde og med redusert dosering i henhold til alder (<60 år: 15 mg/kg, 60-70 år: 12,5 mg/kg og >70 år: 10 mg/kg). Det er også redusert dosering mht. alvorlig nyresvikt (definert som kreatinin > 300 μmol/L eller eGFR ≤ 30 ml/min/1,73m2) der man reduserer doseringen med 2,5 mg/kg som kommer i tillegg til reduksjon i henhold til alder (<60 år: 12,5 mg/kg, 60-70 år: 10 mg/kg og >70 år: 7,5 mg/kg) (Tabell 3)
Tabell 3. Doseendring av intravenøs cyklofosfamid relatert til alder og nyrefunksjon
| Intravenøs cyklofosfamid puls (mg/kg)
(Max 1200 mg/puls) |
||
| Alder (år) | Kreat. ≤ 300(μmol/L) eller
eGFR > 30 (ml/min/1,73m2) |
Kreat. > 300
eGFR: ≤ 30 |
| < 60 | 15 mg/kg | 12,5 mg/kg |
| 60 – 70 | 12,5 mg/kg | 10,0 mg/kg |
| > 70 | 10,0 mg/kg | 7,5 mg/kg |
De første tre doseringene gis med to ukers intervall og neste tre kurer, eller flere, med tre ukers intervall. Uke 0, 2, 4, 7, 10 og 13 og hvis ikke remisjon uke 16, 19 etc. Ved økt benmargshemming må man vurdere å utsette behandling og evt. redusere doseringen. Nadir hematologiske verdier er dag 8-12. Det er viktig med god hydrering og spesial oppmerksomhet ved urinretensjon og å gi behandlingen tidlig om dagen med intravenøs væske behandling. Antiemetisk behandling anvendes og da oftest intravenøs selektiv 5-HT3-reseptorantagonist, for eksempel intravenøs ondansetron 8 mg som kan gjentas ved forsinket kvalme. Ved intravenøs cyklofosfamid behandling gis vanligvis profylaktisk behandling med intravenøs forut og senere to ganger per oral mesna (Uromitexan®) som reduserer dannelsen av akrolein i urinen og minsker risiko for bivirkning fra urinveier som gis forut og to ganger etterpå. Generelt gir man 20% (0,2 x cyclofosfamid doseringen i mg) intravenøst forut og 40% (0,4 x cyclofosfamid doseringen i mg) gjentatt to ganger. For eksempel pasienten som får 900 mg cyklofosfamid får 0,2 x 900 = 180 mg IV Uromitexan® og 0,4 x 900 = 360 mg ≈ 400 mg per oral. Det finnes 400 mg og 600 mg Uromitexan® tabletter som har delstrik.
Per oral cyklofosfamid behandling:
Valg av per oral cyklofosfamid behandling kan vurderes og vanlig doseringen er 2mg/kg gitt tidlig om dagen i en dosering. Sendoxan tabletter er på 50 mg og uten delestrek. Det er liknende dosereduksjon i henhold til alder og nyresvikt som ved intravenøs behandling (Tabell 4)
Tabell 4. Doseendring av per oral cyklofosfamid relatert til alder / nyrefunksjon
| Daglig per oral cyclofosfamid (mg/kg)
(Max 200 mg/dag) |
||
| Alder (år) | Kreat. ≤ 300(μmol/L) eller
eGFR > 30 (ml/min/1,73m2) |
Kreat. > 300
eGFR: ≤ 30 |
| < 60 | 2,0 mg/kg | 1,5 mg/kg |
| 60 – 70 | 1,5 mg/kg | 1,25 mg/kg |
| > 70 | 1,25 mg/kg | 1,0 mg/kg |
Kombinasjonsbehandling med rituximab (RTX) og cyklofosfamid (CYC):
Det er svært begrenset med data på kombinasjonsbehandling med RTX og CYC. ACR retningslinjene for behandling av AAV fra 2021 fraråde den kombinasjonen, basert på at det foreligger ingen studier som påviser bedre effekt av kombinasjon og det er dokumentert økt fare for alvorlige infeksjoner (22). EULAR anbefalingene (23), påpeker manglende dokumentasjon av fordelen ved RTX/CYC kombinasjonsbehandling og påpeker komplikasjoner men men uttrykker seg noe svakere. Det er kolleger som er overbeviste på fordeler av RTX/CYC kombinasjonsbehandling i induksjonsfasen og hevder at det gir (a) raskere effekt en RTX alene og (b) reduserer steroidbehov over tid. Både a og b er foreløpig udokumentert. RTX/CYC kombinasjonsbehandling bidrar til økt infeksjonsrisiko og på sikt sannsynligvis til økt risiko for immunsvikt med hypogammaglobulinemi. I tillegg gir cyclofosfamid økt kreftrisiko. Det pågår en nederlansk studie ut fra universitetssykehuset i Leyden, ENDURRANCE-1, som sammenligner RTX/CYC kombinasjonsbehandling med konvensjonell RTX behandling og forventes avsluttet medio 2025.
I de norske behandlingsretningslinjnene er unntaksvis åpnet for RTX/CYC kombinasjonsbehandling hos livstruende syke pasienter.
C: Tredje valg ved induksjonsbehandling ved GPA:
Kan vurderes unntaksvis ved mindre syke pasienter.
Mykofenolat mofetil (MMF):
Dosering: 2-3 g/d fordelt på to doseringer, med ca. 12 timers intervall. Det er tabletter på 500 mg og kapsler på 250 mg i tillegg finnes MMF i mikstur. Alternativt til MMF er mykofenolsyre/Myfortic (MPA) der doseringen er 360 mg x2 eller 750mg x2. MMA kan ha noe mindre gastrointestinal bivirkninger en MMF men virkningsmekanismen er lik. Generelt vil man ikke anvende MMF som induksjonsbehandling hos de sykeste pasientene. Som induksjonsbehandling hos GPA og MPA pasienter med mindre alvorlig sykdom kommer MMF brukbart ut i hvert fall sammenlignet med cyklofosfamid i en nylig publisert randomisert studie (31). MMF var initialt ikke statistisk dårligere en cyklofosfamid, men induksjonsbehandling med MMF resulterte høyere andel tilbakefall spesielt hos PR3-ANCA antistoff positive AAV pasienter. Som vedlikeholdsbehandling kommer MMF dårligere ut i en randomisert «head-to-head» studie en azathioprin (32).
Metotreksat (MTX):
MTX p.o eller s.c. i ukentlig dosering er medikament som brukes i et stort omfang innen revmatologien noe alle revmatologer er og bør være kjent med. Generelt er MTX s.c. er å foretrekke, der det gir mer stabil blodverdi. Vanlig dosering er opp mot ca. 0,3 mg/kg/uke (15-25 mg) og ofte trapper man gradvis opp mot måldosering over noen uker. Det har vært tradisjon i Europa å gi samtidig folsyre tilskudd for å redusere bivirkninger oftes med 1 mg Folsyre daglig.
En Europeisk studie sammenlignede mindre syke pasienter med MPA og GPA uten nyresvikt (kreatinin < 150 µmol/L) sammenlignet behandling med MTX og cyklofosfamid (CYC). Resultater var at det gikk noe lengre tid å komme i remisjon med MTX og til tross antall av de som kom i remisjon var ikke signifikant dårligere en CYC var det stor forskjell på hyppighet av tilbakefall mellom de to induksjonsbehandlingene. Der var nesten 70% som fikk tilbakefall av de som fikk MTX vs. 47% av de som fikk CYC som induksjonsbehandling etter 18 måneder (33).
Per dags dato kan man vurderer man MTX og evt. MMF som induksjonsbehandling sammen med steroid behandling ved mindre alvorlig form av GPA og MPA. Det foreligger lite data på GPA på kombinasjonsbehandling metotrexat med Rituksimab som brukes i relativ utstrakt grad hos pasienter med rheumatoid artritt.
II. VEDLIKEHOLDSBEHANDLING
Det er foreløpig ikke helt klart hvor lenge vedlikeholdsbehandlingen ved GPA skal vare og sannsynligvis og der må gjøres individuell evaluering. Vedlikeholdsbehandlingen bør i hvert fall vare i minst 2 år, men betydelig del av pasientene trenger behandling adskillig lengre opp til 4-5 år og noen sannsynligvis enda lengre. Det er balanse mellom residivfare av GPA og bivirkninger av langvarig immunsuppresjon og individuell risiko for det. REMAIN studien sammenlignet pasientgruppe med GPA og MPA som hadde vært behandlet med induksjon med cyklofosfamid og prednisolon og i var remisjon på vedlikeholdsbehandling på azathioprin (AZA) samt prednisolon behandling og etter 18-24 måneder fra diagnose og de ble de randomisert til å få videre enten IV rituximab (RTX) 500 mg hver 6. måned eller placebo, der relapsrisiko etter 48 måneder fra diagnose var primært endepunkt. Resultatene var entydig det var nærmere tre ganger flere tilbakefall hos dem som fikk placebo vedlikehold etter 18-24 måned sammenlignet med de som fikk videre rituximab (63% mot 22%, p <0,0001) (37). Den studien viste også at positiv ANCA ved randomisering var assosiert med nærmest dobbelt økt relaps risiko (51%) sammenlignet med de som hadde negativ ANCA status (29%) ved randomisering (p=0.017).
GPA og MPA pasisenter som får induksjonsbehandling med rituksimab (RTX) med god toleranse, anbefalles å fortsette med RTX hver 6. måned. Det er påvist at vedlikeholdsbehandling med RTX er betydelig bedre får å forhindre tilbakefall en konvensjonell azathioprin (AZA) behandling (38). Dette er bl.a. publiserte studier fra GFEV, den franske vaskulitt foreningen i flere studier inkl. MAINRITSAN I, II og III som anvender RTX 500 mg hver 6. måned (38, 39, 40). Dette har vært påvist at RTX 500 mg hver 6. måned etter den franske MAINRITSAN protokoll, at regelmessig dosering bedre en tilpasset (“tailored”) dosering (basert på ANCA status og B-cellesupresjon med CD19 tall) for å hindre tilbakefall. Alternativt kan man anvende vedlikeholdsbehandling med RTX etter RITAZEREM protokollen som anvendte 1000 mg hver 4. måned og viste også betydelig forskjell til fordel av RTX sammenlignet med peroral AZA (2mg/kg/d) på relaps etter totalt 24 måneders behandling med hazzard ratio (HR) 0,41 (95% KI 0,27-0,61, p<0,001). 45% (38/85) av de som fikk RTX hadde relaps mens 71% (60/85) av de som fikk AZA (43).
En tidligere undersøkelser har vist at AZA er overlegen mykofenolat mofetil (MMF) når det gjelder å hindre tilbakefall ved GPA (29), men MMF kan vurderes hvis intoleranse for AZA. Det er forskjellige tradisjoner mellom land i henhold til vedlikeholdsbehandling med lavdose steroider. I flere land i Europa har man brukt lavdose Prednisolon dvs. dosering rundt 5 mg/d over noe lengre tid. Det foregår studier for å se på dette i henhold til bivirkninger og fare for tilbakefall. En amerikansk studie ser på dette kalles TAPIR studien “The Assessment of Prednisone in Remission Trial” og resultater foreligger foreløpig ikke (NCT01933724). I tillegg foregår den franske MAINEPSAN studien “Evaluate the Remission MAINtenance Using Extended Administration of Prednisone in Systemic Anti-neutrophil Cytoplasmic Antibodies Associated Vasculitis” (NCT03290456) som går ut på det samme der resultatene forventes 2024-2025.
Vedr. vedlikeholdsbehandling velger anbefaler både Norsk revmatologisk forening (NRF) og andre inklusiv den franske GFEV å anvende RTX 500 mg hver 6. måned som vedlikeholdsbehandling. I de nylig oppdaterte ACR retningslinjene tar man ikke stilling til vedlikeholdsdoseringen av rituximab og sidestiller 500 mg og 1000 mg hver 6. måned og 1000 mg hver 4. måned, med det argument at det foreligger ingen prospektive studier som sammenlinger disse protokollene (44). Det er dessverre ingen pågående studier som sammenligner disse.
Hvis det ikke er toleranse for RTX på den ene siden at pasienten får allergiske reaksjon og evt. påvisning av anti-RTX antistoffer som kan måles ved OUS eller det at pasienten ikke får klinisk effekt og manglende suprimering av CD19+ B-celler i perifer blod. Vil man i neste omgang vurdere å bruke per oral Imurel(AZA) og deretter evt metotreksat forut mykofenolat mofetil som er eventuelt tredje valg og per oral leflunomid som evt. fjerde valg. Alternativt kan man bruke “off label” anti-CD20 humaniserte antistoffene; obinutuzumab/Gazyvaro® eller okrelizumab/Ocrevus® som er registrerte for bruk av kronisk lymfatisk leukemi (KLL) og follikulært lymfom (FL) og multiple sklerosis (MS).
Azathioprin (AZA) – (Imurel®) doseres oftest rundt 2-2,5 mg/kg i en enkel dosering. Imurel finnes i 50 mg og 25 tabletter og det går ikke å dele dem. Man anbefaler å ta tiopurin methyltransferase (TPMT) genotype forut behandling. De fleste (rundt 90 %) har TPMT*1/*1 (wild-type) genotype med normal TPMT enzym aktivitet. Pasienter som er heterozygote med TPMT*1 og noen av de over 20 TPMT polymorfismeme, som oftest er *2, *3A, *3B, *3C, og *4, har redusert TPMT enzym aktivitet og må få redusert, oftest halvert dosering og følges grundig hvis oppstart. De som ikke har TPMT*1genotypen og enten homozyg og eller heterozygot for en av de allelene bør ikke få azathioprin i hele tatt pga. økt risiko for alvorlig myelosuppresjon. Dette utgjør få pasienter (>1%). Vises til Avdeling for farmakologi. Oslo universitetssykehus.
Verdi av 6-tioguaninnukleotide (6-TGN) og metyl-merkaptopurin (me-MP) måles i heparinisert fullblod, tilsier om effekt av AZA behandlingen. Terapeutisk område er: 6-TGN 3,5−5,0 µmol/L og me-MP <50 µmol/L.
Dette er et relativt stort problem ved immunsuppressiv behandling av GPA og bor behandles aggressivt. Ved langvarig immunsuppresjon anbefales profylaktisk behandling for Pneumocystis jiroveci pneumoni (PJP). Det finnes forskjellige alternativer for forebyggende behandling. Oftest anvedes per oral Bactrim (SMZ-TMP) (400 mg sulfamethoxazol og 80 mg trimetoprim) daglig. Ved allergi/intolleranse kan man anvende Dapson 50-100 mg/d, det er en viss fare for krysssulfaallergi. Det tredje alternativet er pentamidin inhalasjon enten en gang i måneden (300 mg) eller hver annen hver uke (150 mg). Det er sjelden alvorlige bivirkninger med inhalasjonsbehandling. Men den kan ofte gi hoste, bronkospasme, samt og smaksforandringer og uvelfølelse.
–Høy alder og lungesykdom. Det er økt risiko for PJP når pasienten tidligere har hatt PJP, økt alder over 65 år, påvist lungesykdom eller omfattende lungeaffeksjon av AAV, ved høy glukokortikoid dosering særlig ved Prednisolon over 20 mg/d og også hos pasienter med redusert CD4+ T-lymfocytt-tall særlig hos de som ligger under 200 x10E6 celler/liter. Flere risikofaktorer styrker behandlingsindikasjonen ytterligere.
-Hypogammaglobulinemi kan sees ved AAV og det er definert flere risikofaktorer bl.a. lav IgG forut start og cyklofosfamid behandling før eller med rituksimab behandling. Det er assosiasjon mellom dose og lengde av RTX behandling og hypogammaglobulinemi. Det anbefales at man kontroller gammaglobulinkonsentrasjon ved oppfølging. Moderat hypogammaglobulinemi er ofte definert hos de som har IgG 3,0 – 4,9 g/L og alvorlig de som har IgG < 3,0 g/L. Hvis det foreligger i tillegg økt infeksjonsrisiko er det klar indikasjon for substitusjonsbehandling med sc. eller iv. gammaglobulin substitusjon.
Svangerskap og fødsel
Det er begrenset erfaring på svangerskap og AAV. Det er anført vellykket svangerskap relatert til rituksimab behandling til tross det ikke har vært formelt vurdert med henhold til dette. De to andre behandlingsalternativene ved alvorlig AVV er per oral eller intravenøs cyklofosfamid, mykofenolat mofetil og evt. metotreksat, men alle de er knyttet opp til teratogenitet. Azathioprin kan brukes under svangerskap, men det er mer egnet som vedlikeholdsbehandling. Det er rapporter om vellykket svangerskap der den kommende mor har fått cyklofosfamid behandling i tredje trimester, i tillegg der pasienten har fått plasmaferese og intravenøs immunglobulin i tillegg til steroid behandling. Nylig publisert metaanlyse inkluderte åtte studier, med detaljer om 82 graviditeter hos 64 kvinner (42). De vanligste legemidlene som ble brukt til remisjonsinduksjon før unnfangelse var cyklofosfamid, rituximab, prednisolon og azatioprin. Alvorlige morskomplikasjoner i svangerskapet inkluderte progressiv trakeal/subglottisk stenose (n=5), nyresykdom (n=2), preeklampsi (n=10) og spontanaborter (n=5). Fetale anomalier var sjeldne (n=5). Gjennomsnittlig fødselsvekt var 3,37 kg og gjennomsnittlig svangerskapsalder var 38,3 uker. Ingen mødredødsfall eller vaskulitt hos nyfødte ble rapportert. Forfatterende konklulderte at AAV pasienter har som oftest positivt utfall for både mor og fosterutfall til tross for kraftig induksjonsterapi. Det er viktig med overvåking og rask behandling av evt oppbluss av AAV under graviditet. Alvorlige komplikasjoner og oppbluss var ikke assosiert med dårligere utfall for de nyfødte (42). Viser også fra informasjon fra Nasjonal kompetansetjeneste for svangerskap og revmatiske sykdommer (NKSR) ved St. Olavs universitetssykehuset i Trondheim.
Prognose
Det er økende overlevelse ved GPA som ved de andre AAV mens prognosen er svært dårlig ved systemisk GPA uten behandling. Data her fra Norge fra Norsk systemisk bindevevssykdom og vaskulitt-register (NOSVAR) sammenlignet med normalbefolkning viste at samlet sett var 91% 5 års overlevelse og 80% 10 års overlevelse hos norske AAV pasienter sammenlignet med 95% og 87% hos alders og kjønns matchet friske kontroller. Standardisert dødelighet rate var 1,5 (95% konfidensintervall var 1,0-2,1) (42). Økt risiko for dødelighet er nyresvikt, som og økt alder men affeksjon av tarm og hjerte i tillegg til lungeblødninger i akutt fasen. Både aktivitet av grunnsykdommen og infeksjonsrelaterte komplikasjoner som kan både være assosiert til sykdom og behandlingen er hoveddødsårsakene. Man forventer at risikoen for cancerrelaterte komplikasjoner går ned med mindre utstrakt bruk av cyclofosfamid i induksjonsbehandling.
Største problemet med GPA er tilbakefall. Generelt er 5 år tilbakefall fri overlevelse på 30-40%, men man forventer at dette blir opp mot 60-70% med langvarig rituksimab vedlikeholdsbehandling (43). Det er svært viktig med langvarig oppfølging får å fange opp tilbakefall.
Anbefalt litteratur
- Kronbichler, A., et al. (2024). “Diagnosis and management of ANCA-associated vasculitis.” Lancet 403(10427): 683-698.
- Wallace ZS, Miloslavsky EM. Management of ANCA associated vasculitis. BMJ 2020;368:m421.
- Trivioli, G., et al. (2022). “Genetics of ANCA-associated vasculitis: role in pathogenesis, classification and management.” Nat Rev Rheumatol 18(10): 559-574
- Kitching AR, et al. ANCA-associated vasculitis. Nature Reviews Disease Primers. 2020;6(1):71.
- Bokkapittel: Pagnoux C. Granulomatosis with Polyangiitis. In: Sinico RA, Guillevin L, editors. Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis. Cham: Springer International Publishing; 2020. p. 97-129
Referanser
- Lyons PA. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367:214-23.
- Lyons PA, Peters JE, Alberici F, Liley J, Coulson RMR, Astle W, et al. Genome-wide association study of eosinophilic granulomatosis with polyangiitis reveals genomic loci stratified by ANCA status. Nat Commun. 2019;10(1):5120.
- Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of Functional and Expression Polymorphisms Associated With Risk for Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis. Arthritis & rheumatology (Hoboken, NJ). 2016.
- Xie G, Roshandel D, Sherva R, Monach PA, Lu EY, Kung T, et al. Association of granulomatosis with polyangiitis (Wegener’s) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum. 2013;65(9):2457-68.
- Pearce FA, Craven A, Merkel PA, Luqmani RA, Watts RA. Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology (Oxford). 2017;56(11):1962-9.
- Mahr A, Katsahian S, Varet H, Guillevin L, Hagen EC, Höglund P, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-10.
- Holle J, Gross W, Latza U, Nolle B, Ambrosch P, Heller M, et al. Improved outcome of 445 Wegener’s granulomatosis patients in a German vasculitis center over four decades. Arthritis Rheum. 2010;63:257 – 66.
- Holle J, Gross W, Holl-Ulrich K, Ambrosch P, Noelle B, Both M, et al. Prospective long-term follow-up of patients with localized Wegener’s granulomatosis: does it occur as persisten disease stage? Ann Rheum Dis. 2010;69:1934 – 9.
- Maillet T, Goletto T, Beltramo G, Dupuy H, Jouneau S, Borie R, et al. Usual interstitial pneumonia in ANCA-associated vasculitis: A poor prognostic factor. J Autoimmun. 2020;106:102338.
- Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628-36.
- Merkel PA, Lo GH, Holbrook JT, Tibbs AK, Allen NB, Davis JC, Jr., et al. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005;142(8):620-6.
- Lillejordet E, Rashid HU, Evang JA, Taraldsrud E, Gunnarsson R. A woman in her forties with influenza symptoms and swollen arm. Tidsskr Nor Laegeforen. 2019;139(1).
- Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33(8):1101-7.
- Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007;66(2):222-7.
- Robson JC, Grayson PC, Ponte C, Suppiah R, Craven A, Judge A, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis. 2022;81(3):315-20.
- Suppiah R, Robson JC, Grayson PC, Ponte C, Craven A, Khalid S, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis. 2022;81(3):321-6
- Grayson PC, Ponte C, Suppiah R, Robson JC, Craven A, Judge A, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann Rheum Dis. 2022;81(3):309-14.
- Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87(11):671-8.
- Mukhtyar C, Lee R, Brown D, Carruthers D, Dasgupta B, Dubey S, et al. Modification and validation of the Birmingham Vasculitis Activity Score (version 3). Ann Rheum Dis. 2009;68(12):1827-32.
- Exley A, Bacon P, Luqmani R, Kitas G, Gordon C, Savage C, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997;40:371 – 80.
- Terrier B, Charles P, Aumaître O, Belot A, Bonnotte B, Crabol Y, et al. ANCA-associated vasculitides: Recommendations of the French Vasculitis Study Group on the use of immunosuppressants and biotherapies for remission induction and maintenance. Presse medicale (Paris, France : 1983). 2020;49(3)
- Chung SA, Langford CA, Maz M, Abril A, Gorelik M, Guyatt G, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2021;73(8):1366-83.
- Hellmich B, Sanchez-Alamo B, Schirmer JH, Berti A, Blockmans D, Cid MC, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2024;83(1):30-47.
- Walsh M, Merkel PA, Peh CA, Szpirt WM, Puechal X, Fujimoto S, et al. Plasma Exchange and Glucocorticoids in Severe ANCA-Associated Vasculitis. N Engl J Med. 2020;382(7):622-31.
- Walsh M, Collister D, Zeng L, Merkel PA, Pusey CD, Guyatt G, et al. The effects of plasma exchange in patients with ANCA-associated vasculitis: an updated systematic review and meta-analysis. Bmj. 2022;376:e064604.
- Stone J, Merkel P, Spiera R, Seo P, Langford C, Hoffman G, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221 – 32.
- Jones R, Tervaert J, Hauser T, Luqmani R, Morgan M, Peh C, et al. Rituximab versus cyclophopshamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211 – 20.
- Bénard V, Farhat C, Zarandi-Nowroozi M, Durand M, Charles P, Puéchal X, et al. Comparison of Two Rituximab Induction Regimens for Antineutrophil Cytoplasm Antibody-Associated Vasculitis: Systematic Review and Meta-Analysis. ACR Open Rheumatol. 2021;3(7):484-94.
- Unizony S, Villarreal M, Miloslavsky EM, Lu N, Merkel PA, Spiera R, et al. Clinical outcomes of treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis based on ANCA type. Ann Rheum Dis. 2016;75(6):1166-9.
- de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670-80.
- Jones RB, Hiemstra TF, Ballarin J, Blockmans DE, Brogan P, Bruchfeld A, et al. Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA-associated vasculitis: a randomised, non-inferiority trial. Ann Rheum Dis. 2019;78(3):399-405.
- Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. 2010;304(21):2381-8.
- De Groot K, Rasmussen N, Bacon P, Tervaert J, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005;52:2461 – 9.
- Jayne DRW, Merkel PA, Schall TJ, Bekker P, Group AS. Avacopan for the Treatment of ANCA-Associated Vasculitis. The New England journal of medicine. 2021;384(7):599-609.
- Cortazar FB, Niles JL, Jayne DRW, Merkel PA, Bruchfeld A, Yue H, et al. Renal Recovery for Patients with ANCA-Associated Vasculitis and Low eGFR in the ADVOCATE Trial of Avacopan. Kidney Int Rep. 2023;8(4):860-70.
- Cortazar FB, Cerda J, Dhanani R, Roglieri J, Santoriello D. Avacopan in Patients With Rapidly Progressive Glomerulonephritis Requiring Dialysis. Kidney Int Rep. 2023;8(8):1687-91.
- Karras A, Pagnoux C, Haubitz M, Groot K, Puechal X, Tervaert JWC, et al. Randomised controlled trial of prolonged treatment in the remission phase of ANCA-associated vasculitis. Ann Rheum Dis. 2017;76(10):1662-8.
- Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaitre O, Cohen P, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771-80.
- Charles P, Terrier B, Perrodeau E, Cohen P, Faguer S, Huart A, et al. Comparison of individually tailored versus fixed-schedule rituximab regimen to maintain ANCA-associated vasculitis remission: results of a multicentre, randomised controlled, phase III trial (MAINRITSAN2). Ann Rheum Dis. 2018;77(8):1143-9.
- Charles P, Perrodeau E, Samson M. Long-Term Rituximab Use to Maintain Remission of Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. (MAINRITSAN3) Annals of Internal Medicine. 2020;173(3):179-87.
- Terrier B, Pagnoux C, Perrodeau E, Karras A, Khouatra C, Aumaitre O, et al. Long-term efficacy of remission-maintenance regimens for ANCA-associated vasculitides. Ann Rheum Dis. 2018;77(8):1150-6.
- Ashok, A., et al. (2023). “Incidence, clinical features, management and outcomes of ANCA-associated vasculitis in pregnancy- a systematic literature review.” Sarcoidosis Vasc Diffuse Lung Dis 40(4): e2023040.
- Smith RM, Jones RB, Specks U, Bond S, Nodale M, Al-Jayyousi R, et al. Rituximab versus azathioprine for maintenance of remission for patients with ANCA-associated vasculitis and relapsing disease: an international randomised controlled trial. Ann Rheum Dis. 2023;82(7):937-44.
- Ashok A, Russell L, Dey M, Kouranloo K. Incidence, clinical features, management and outcomes of ANCA-associated vasculitis in pregnancy- a systematic literature review. Sarcoidosis, vasculitis, and diffuse lung diseases. 2023;40(4):e2023040.
- Haris A, Dolgos S, Polner K. Therapy and prognosis of ANCA-associated vasculitis from the clinical nephrologist’s perspective. Int Urol Nephrol. 2017;49(1):91-102.
- Garen T, Lerang K, Hoffmann-Vold AM, Andersson H, Midtvedt O, Brunborg C, et al. Mortality and causes of death across the systemic connective tissue diseases and the primary systemic vasculitides. Rheumatology (Oxford). 2019;58(2):313-20.
- Pagnoux C. Granulomatosis with Polyangiitis. In: Sinico RA, Guillevin L, editors. Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis. Cham: Springer International Publishing; 2020. p. 97-129.
- Kidney Disease: Improving Global Outcomes, A. V. W. G. (2024). “KDIGO 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis.” Kidney Int 105(3S): S71-S116.
