ANDRE SYKDOMMER (REV 063-REV 077)
177 Loeys-Dietz syndrom (REV 063)
Øyvind Palm
- Karpaltunnelsyndrom, Bakers cyste, entesopati, lumbago, isjas, peritendinitt og kapsulitt i skulder er omtalt i egne kapitler.
ICD-10: Q87.4 (samme som Marfans syndrom)
Definisjon
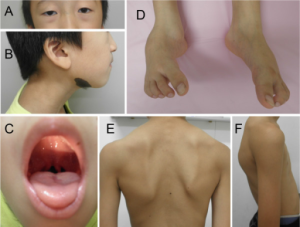
Loeys-Dietz syndrom er en autosomal dominant arvelig sykdom i bindevev som disponerer for aneurismer i aorta og mindre arterier allerede fra barnealder. Sykdommen kan ligne på Marfans syndrom. Sammen med vaskulær form av Ehlers-Danlos syndrom (som angriper blodårer) og hereditært thorakalt aortaaneurisme (HTAD) (Isselbacher EM, 2016), inngår sykdommen blant de arvelige ”Marfan-lignende” sykdommene. Loeys-Dietz syndrom er derfor ikke en autoimmun bindevevssykdom. Genetiske tester har avdekket mutasjoner og flere former av Loeys-Dietz syndrom. Mange har hatt familiemedlemmer som brått døde i ung alder (Gouda P, 2022).
Revmatologens rolle er å skille sykdommen fra vaskulitt og ulike muskelskjelett sykdommer, noe som er av betydning fordi riktig oppfølging av Loeys-Dietz syndrom og operasjon om nødvendig kan forebygge alvorlige komplikasjoner og tidlig død. I Norge er Sunnås sykehus kompetansesenter.
Historie
Sykdommen ble første gang beskrevet i 2005 (Loeys BL, 2005)
Forekomst
Loeys-Dietz syndrom defineres som en sjelden sykdom. Definisjon av sjeldenhet: færre enn ett tilfelle per 2.000 personer (Det Norske Helse og Omsorgs-departementet). Spesifikke data for insidens og prevalens foreligger ikke, men Type 1 og type 2 (se nedenfor) er vanligst.
Sykdomsårsak
Mutasjoner i ulike gener forårsaker sykdommen og subgruppene: Type 1 (GFBR1), type 2 (TGFBR2), type 3 (SMAD3), type 4 (TGFB2,) og type 5 (TGFB3). Likevel påvises sykdommen oftest blant personer uten en familie-historie.
Symptomer
Sykdommen har mange ulike uttrykksformer, til dels avhengig av hvilken av de fem subtypene som genetisk foreligger. Kardiovaskulære-, muskelskjelett-, nevrologiske- og gastrointestinale symptomer er typisk. I noen tilfeller utvikles påfallende misdannelser i ansiktsskjelett og hodeskalle og hudforandringer. Aneurismeutvikling er ofte asymptomatisk. Sammenlignet med Marfans syndrom ses disseksjoner noe oftere ved Loeys-Dietz syndrom (Gouda P, 2022).
Utredning
Klinisk. Det skjer en for tidlig sammenvoksing av skalleben i spedbarnsalder. Pasientene har ofte utvikler en karakteristisk ansiktsform med vid avstand mellom øyne. I tillegg ses ofte uvula bifida (spaltet i drøvel) og ganespalte. I columna ses skoliose eller kyfose. Komplikasjoner med aneurismer i aorta og andre arterier er mest alvorlig. Thoraks med brystben kan utvikle deformiteter i oppveksten. Noen har en “Marfanoid habitus” med lange armer, ben, fingre og tær i forhold til kropp. Forekomsten og alvorlighetsgraden av aneurismer (med ruptur og blødningsrisiko) er individuelt forskjellig, men kan oppstå i barne-årene (Hyung-Tae S, 2015).
Laboratorium. Normale inflammasjonsparametere (utenom ved aneurismeblødning) og normale andre sykdomsmarkører. Genetiske tester: se genetikkportalen.no.
Bildediagnostikk: -MR-angiografi fremstiller de viktigste pulsårene fra og med hodet, halsen, brystet, mageområdet og bekkenet. -Ultralyd Doppler / ekkokardiografi kan bestemme diameter på hovedpulsåren ved hjertet og aortabuen måles. -CT-undersøkelse kan fremstille hele hovedpulsåren. Undersøkelsen gjøres ikke for ofte på grunn av røntgenstråler
Diagnose
Familie-anamnese, symptomer, habitus og påvisning av aneurismer i ung alder. Genetiske tester (genetikkportalen.no)
Differensialdiagnoser
Differensialdiagnoser andre “Marfan-lignende” sykdommer: Marfans syndrom, Ehlers-Danlos syndrom, andre former for arvelige bindevevssykdommer (HTAD med flere) som også kan medføre aortadilatasjon og aneurismer.
Komplikasjoner
De mest alvorlige er aortaaneurismer, oftest ascendens og i aortabuen. Aortadisseksjon (Gouda P, 2022).
Behandling
Ingen helbredende behandling er tilgjengelig. Dersom ett eller flere aneurismer øker i størrelse, kan operasjon for å forebygge ruptur bli nødvendig. Karkirurger / thoraks-kirurger har retningslinjer for når operasjoner bør gjøres.
Blodtrykksmålinger er viktige for å unngå hypertoni som øker risikoen for aortaaneurismer og rupturer.
Vanlig fysisk aktivitet anbefales, mens intensiv, hard fysisk trening øker trykket på blodårene og bør unngås.
Kortison og annen immundempende behandling har ingen plass i behandlingen av arvelige bindevevssykdommer, til forskjell fra autoimmune bindevevssykdommer.
Oppfølging
Regelmessig kontroll av blodårene er viktig for å oppdage aneurismer tidlig. Hvis pulsåren utvider seg, gjøres målinger minst hvert halvår, ellers årlig i en lengre periode
Litteratur
- Gouda P, 2022
- Yarate YA, 2015 (Aortadilatasjon, utvidelse av hovedpulsåren) blant barn og unge
- MacCarric G, 2014 (Dagose og management)
- Loeys BL, 2005 (original beskrivelse av syndromet)
- Kompetansesenter: Sunnås Sykehus
