ANDRE SYKDOMMER (REV 063-REV 077)
183 Metabolske og endokrine tilstander med revmatologiske symptomer, elektrolytter, syre-base, lipider (REV 073, REV 106, REV 107, REV 136, REV 162, REV 163, REV 165, REV 166, REV 167)
Metabolske sykdommer i revmatologi
Jan Tore Gran and Øyvind Palm
Definisjon. Metabolske sykdommer, også kalt stoffskiftesykdommer, er en gruppe tilstander som påvirker kroppens evne til å omsette og bryte ned næringsstoffer. Dette kan føre til en rekke problemer, inkludert ansamling av toksiner, energimangel og vevsskader.
Symptomer. Flere av disse sykdommene kan forårsake revmatiske symptomer, som leddsmerter, stivhet og hevelse. Symptomene varierer, avhengig av sykdomsmekanismen.
Noen autoimmune sykdommer, som for eksempel revmatoid artritt og systemisk lupus erythematosus (SLE), kan være assosiert med metabolske forstyrrelser. Også revmatologiske medisiner, som for eksempel kortikosteroider, kan ha bivirkninger som kan føre til metabolske forstyrrelser.
Revmatologens rolle. Det er av betydning at revmatologer har en god forståelse av metabolske sykdommer for å kunne spille en viktig rolle i diagnosen, behandlingen og oppfølgingen av disse pasientene.
Nedenfor er aktuelle tilstander listet alfabetisk:
Addison sykdom (binyrebarksvikt)
Læringsmål REV 164.Revmatologen skal ha kunnskap om symptomer/tegn ved akutt binyrebarksvikt (Addisons sykdom). Beherske initial behandling av akutt binyrebarksvikt og Addison-krise.

-Symptomer på primær eller sekundær binyrebarksvikt er slapphet/tretthet, generell svakhet og muskel/skjelettsmerter, redusert appetitt, vekttap, kvalme, oppkast, abdominalsmerter, postural hypotensjon/svimmelhet (viktig å måle ortostatisk blodtrykk), salthunger, hyperpigmentering av hud og slimhinner (veldig vanlig), tap av libido hos kvinner, psykiatriske symptomer (forvirring, depresjon, psykose) og eosinofili. Hyponatriemi er vanlig, ofte også hyperkalemi, hyperkalsemi, hypoglykemi, transaminase-stigning og eosinofili kan forekomme. Ved akutt binyrebarksvikt sees hypotensjon/sirkulasjonskollaps, abdominalsmerter, redusert appetitt, kvalme, oppkast, forvirring, feber, hypoglykemi. Hyperpigmentering kan mangle dersom binyrebarksvikten har oppstått raskt (blødning og infarkt i binyrene).
Før behandlingsstart er det ønskelig (men ikke avgjørende i akutte tilfeller) å ta hormonprøver (ACTH, kortisol, aldosteron og renin-aktivitet). Medikamentelt brukes hydrokortison (Solu-Cortef) 100 mg i.v. over 0,5-1 minutt, og deretter 50 mg hydrokortison hver 6. time første døgn. I tillegg gis væske (Nasjonal veileder i endokrinologi, 28.09.2021).
Akromegali
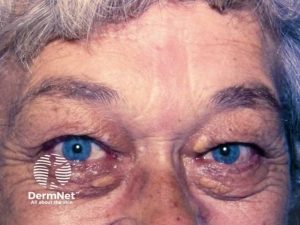
Akromegali skyldes for høy produksjon av veksthormon, oftest på grunn av adenom i hypofysen. Ereksjonssvikt og menstruasjonsforstyrrelser kan være tidlige symptomer. Leddsmerter og karpal tunnel syndrom er blant revmatiske symptomer. Hodepine og bi-temporal hemianopsi, vekstøkning i kjever og panne, tykke øyelokk, stor nese og underleppe er blant klassiske kjennetegn.
Patogenese: Skyldes hyperplasi eller adenom i hypofyse med forstyrret veksthormonproduksjon (Adigun OO, 2023).
Revmatiske manifestasjoner
- Artritt, intermitterende
- Artrose og hypermobilitet hos 75 %.
- Karpal tunnel syndrom bilateralt hos 30-50 %.
- Kondrokalsinose hyppig.
- Leddbrusk, generell hyperplasi
- Myopati med dominerende svakhet av proksimal muskulatur.
- Raynauds-liknende fenomener hos 30 %.
- Ryggsmerter hos 50 %.
Alkaptonuri (Okronose)
Alkaptonuri er en genetisk autosomal recessiv sykdom der mangel på enzymet homogentisinsyre oksidase (HGO) medfører akkumulering av homogentisinsyre. Okronose betegner pigmentering av brusk og deler av bindevev. Sykdommen medfører artrose i store ledd og columna med intravertebral klassifikasjon. Pasientene rapporterer ofte om mørk urin (Sharabi AF, 2023)
Beinsykdommer
Læringsmål REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Beinsykdommer er omtalt i ulike kapitler: Osteoporose, osteopeni og osteomalasi; Paget sykdom: Tumorer i ledd og leddnære strukturer og osteopetrose.
Diabetes mellitus og revmatiske manifestasjoner
- Vennligst se eget kapittel om diabetes mellitus og diabetisk hånd
- Vennligst les om hypoglykemi, hyperglykemi og diabetisk ketoacidose i kapitlet om diabetes mellitus.
Elektrolytt-forstyrrelser
Læringsmål REV 106. Revmatologen skal under supervisjon kunne diagnostisere forstyrrelser i væske- og elektrolyttbalansen, og initialt behandle med intravenøs væske, diuretika og elektrolytter. Ha kunnskap om følgende forstyrrelser: hyponatremi, inklusive kronisk hyponatremi, hypernatremi, hypokalemi, hyperkalemi, hypokalsemi, hyperkalsemi, hypofosfatemi, hypomagnesemi.
Læringsmål REV 136. revmatologen skal ha kunnskap om de metabolske komplikasjonene ved nyresykdom, herunder elektrolyttforstyrrelser, syre-/baseforstyrrelser og forstyrrelser i mineralmetabolismen
Definisjon. Elektrolytter er nødvendige for cellenes funksjon, inklusiv å utløse aksjonspotensial i nerver og muskelceller. Natrium, kalium og klorid er sammen med magnesium, kalsium, fosfat og bikarbonat essensielle elektrolytter som stammer fra inntak av mat og væske.
Litteratur: Scrimanger I, 2023
- Syre-baseforstyrrelser er omtalt nedenfor i dette kapitlet.
Fenylketonuri
Fenylketonuri (PKU) er en arvelig stoffskiftesykdom som fører til opphopning av aminosyren fenylalanin i blodet. Ubehandlet kan dette gi alvorlige konsekvenser som utviklingshemming, epilepsi og andre nevrologiske problemer (van Spronsen FJ, 2021).
Feokromocytom
Feokromocytom er en tumor som utgår fra kromaffine celler i binyremargen eller i paraganglion celler. Omtrent 90% er sporadiske, mens 10% er assosiert til bakenforliggende genetiske sykdommer som Von Hippel–Lindau syndrom, type 1 neurofibromatose og multiple endokrin neoplasi syndrom type IIA og type IIB (Bryant J, 2003). Symptomer skyldes økt katekolamin produksjon og omfatter symptomfrihet, hodepine (50%), hjertebank (60%), svette (50%), hypertensiv krise, blekhet, tremor, utmattelse, angst, feber, smerte og flushing (20%) (Lee TH, 2002).
Utredning omfatter måling av Plasma-metanefrin/p-normetanefrin i plasma. Urinanalyser av mefanefrin eller vanillinmandel-syre etter måling i 24 timer samling er mindre sensitive (Nasjonal veileder, endokrinologi, 28.09.2021). Behandlingen er fortrinnsvis kirurgisk reseksjon (Mubarik A, 2023).
Hemokromatose
Læringsmål REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst les om hemokromatose i eget kapittel
Hypo-, hyperglykemi og ketoacidose
Læringsmål REV 162. Revmatologen skal beherske diagnostikk og behandling av hypoglykemi, hyperglykemi og diabetisk ketoacidose.
Læringsmål REV 163. Revmatologen skal ha god kunnskap om behandling av non-ketotisk hyperosmolær koma.
- Vennligst les om hypoglykemi, hyperglykemi og diabetisk ketoacidose i kapitlet om diabetes mellitus.
Hyperlipidemi
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
. CC BY-NC 4.0")
EULAR anbefaler screening for kardiovaskulær sykdom regelmessig (minst hvert 5. år) ved revmatoid artritt. R Agca, et al 2016. Også SLE, Takayasus arteritt og mange andre inflammatoriske revmatisk sykdommer er beheftet med økt risiko for aterosklerose. En vesentlig risiko er samtidig hyperlipidemi som krever optimal behandling.
Klassifikasjon av Hyperlipidemier
- Primære Dyslipidemier: –Polygent betingede. -Monogent betingede
- Sekundære Dyslipidemier
–Artralgier og artritt kan sees ved type II a og en sjelden gang ved type IV
-Xantomer lokalisert til sever sees ved type II a og III
–Urinsyregikt assosiert med hypertriglyseridemi ved type I, IV og V hyperlipoproteinemi
Generelt om dyslipidemier. Blodets lipider opptrer alltid bundet til proteiner (lipoproteiner – LP). LP er makromolekyler som transporterer både kolesterol og triglyserid. De er oppbygde med en fettholdig kjerne og en kappe som består av fosfolipider og apolipoproteiner. Apo LP sørger for binding til cellemembraner og inngår som co-faktorer for enzymer. ApoB finnes i LDL (Low Density Lipoprotein), apoA-1 og apoA-2 finnes i HDL (High Density Lipoprotein). Fettet transporteres i sirkulasjonen som lipider – enten som frie fettsyrer (fra fettvev) eller som lipoproteiner (fra tarm og lever).
Kolesterol: 2/3 er bundet til LDL. Høye verdier øker kalsium inn-fluks i arterienes glatte muskelceller som får økt tonus og motstand i kar. Risiko for død av iskemisk hjertesykdom øker med økende serumnivå. Også risikoen for cerebrovaskulær sykdom synes a øke.
Triglyserider: Økte mengder er forbundet med moderat økt risiko for hjerte/karsykdom. Økte serumkonsentrasjoner henger ofte sammen med lavt HDL-kolesterol.
HDL-kolesterol: Lave konsentrasjoner er assosiert til iskemisk hjertesykdom uavhengig av mengden total-kolesterol. Et inntrykk av konsentrasjonen kan også fås ved måling av apolipoprotein A1 (apoA-1).
Lipoprotein (a): Lipoprotein “lille a” er et LDL og høye konsentrasjoner gir økt risiko for iskemisk hjertesykdom. Ca 5% av befolkningen har forhøyet verdi, oftest av genetiske årsaker. Lett forhøyet er Lp(a) 75-125 nmol/L (300-500 mg/L), mes sterkt forhøyet er >400 nmol/L (>1800 mg/L). Inntil spesifikke Lp(a)-senkende medikamenter er tilgjengelige på markedet, er generelle forebyggende tiltak aktuelt (Svilaas T, 2023).
Svær hyperkolesterolemi: Defineres som serumkonsentrasjon > 13mmol/L. Disse pasientene har også et betydelig forhøyet LDL kolesterol, mens HDL kolesterol-nivået er normalt eller redusert.
Kombinert hyperlipidemi: Har foruten svær hyperkolesterolemi også høye triglyseridverdier (> 10mmol/L). Disse er ofte sekundære til cholestatisk leversykdom, nefrotisk syndrom, hypothyreose, alkoholmisbruk, diabetes og bulemi.
Utredning: Total-kolesterol, HDL-kolesterol, triglyserid tatt fastende, LDL-kolesterol beregnes ifølge. Friedewalds formel: totalkolesterol – HDL-kolesterol – 0,45 x triglyserider (mmol/I)
Behandlingsmål: For å redusere risikoen for hjertekarsykdom er det hensiktsmessig å senke konsentrasjonen av kolesterol og/eller triglyserid, og hvis mulig øke konsentrasjonen av HDL (lavt HDL forhindrer normal fjernelse av kolesterol fra karveggen). Kolesterol holdes under 5,0 mmol/L og LDL-kolesterol under 3,0 mmol/1 eller lavest mulig. Ved koronar-sykdom er et behandlingsmål LDL under 1,4 mmol/L.
To typer av hyperlipidemi kan sees ved SLE. Den ene typen opptrer ved aktiv sykdom og da særlig hos barn. Den karakteriseres av lav HDL, lav apo-A1 og høye verdier av VLDL og triglyserid. Den andre typen sees ved behandling med kortikosteroider. Disse medikamentene virker direkte koronarskadelige ved å øke kolesterol, triglyserid, blodtrykket, serum glukose, vektøkning og homocysteinemi – kanskje også direkte på karene. Homocystein har direkte toksiske effekter på endotel. Både hyperkolesterolemi, hypertriglyseridemi og hyperhomocysteinemi sees relativt ofte ved SLE, og den økte risikoen for koronarsykdom bør forebygges. (Homocystein er en aminosyre som dannes fra metionin, en annen aminosyre som opptrer i alle proteiner for det omdannes til cystein. Kjente årsaker til forhøyet homocystein er mangel på folat, vit B6 og vit B12. Supplerende litteratur: Hill MF, 2023.
Hyperparathyroidisme og Hypoparathyroidisme
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst se eget kapittel om paratyreoidea-sykdommer
Lesch-Nyhans syndrom
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
Lesch-Nyhans syndrom (Michal Lesch 1938-2008 og William Nyhan f 1926) er en X-kromosomal recessiv genetisk metabolsk sykdom som påvirker purin-metabolismen. Sykdommen medfører høye nivåer av urinsyre i blodet. Ubehandlet ses nyrestener, nyresvikt, og urinsyregikt (Bell S, 2016).
Vennligst se også Purinmetabolisme nedenfor i dette kapitlet.
Lipodystrofi
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
. CC BY 3.0")
Definisjon: Lipodystrofi er enten genetisk eller ervervet og defineres ved feil distribusjon av fett i kroppen. Det finnes begrensede og generaliserte former. Sykdommen er progressiv og kan være en komplikasjon til revmatiske sykdommer. Lipodystrofi er sjelden og opptrer på ulike måter. Sykdommen blir derfor ofte feildiagnostisert eller oversett selv om noen forløp kan være alvorlige (Araújo-Vilar D, 2019).
| Inndeling av lipodystrofier (tabell modifisert etter Araújo-Vilar D, 2019). |
| 1 Kongenitale former |
| 1.1 Generalisert (Berardinelli-Seip syndrom) |
| 1.1.1 Type 1 Kongenital generalisert lipodystrofi (AGPAT2, recessiv, OMIM #608594) |
| 1.1.2 Type 2 Kongenital generalisert lipodystrofi (BSCL2, recessiv, OMIM #269700) |
| 1.2.3 Type 3 Kongenital generalisert lipodystrofi (CAV1, recessiv, OMIM #612526) |
| 1.2.4 Type 4 Kongenital generalisert lipodystrofi (PTRF, recessiv, OMIM #613327) |
| 1.2.5 PPARG-assosiert Kongenital generalisert lipodystrofi (recessiv) |
| 1.2 Partial/begrenset |
| 1.2.1 Type 1 Familiær partial lipodystrofi (Köbberling syndrom; ukjente gener, dominant eller polygen, OMIM #608600) |
| 1.2.2 Type 2 Familiær partial lipodystrofi (Dunnigan disease; LMNA, dominant or codominant, OMIM #151660) |
| 1.2.3 Type 3 Familiær partial lipodystrofi (PPARG, dominant, OMIM #604367) |
| 1.2.4 Type 4 Familiær partial lipodystrofi (PLIN1, dominant, OMIM #613877) |
| 1.2.5 Type 5 Familiær partial lipodystrofi (CIDEC, recessive, OMIM #615238) |
| 1.2.6 Type 6 Familiær partial lipodystrofi (LIPE, recessive, OMIM #615980) |
| 1.2.7 AKT2-linked lipodystrofi (dominant) |
Epidemiologi: Når HIV-relatert lipodystrofi holdes utenom er prevalens beregnet til 3/million. Begrenset lipodystrofi er vanligst (prevalens 2,4/mill), mens generalisert type bare utgjør 0,23/million (Chiquette E, 2017).
Sykdomsårsak: Familiær partiell lipodystrofi (FPL) skyldes mutasjoner av Lamin A-genet (lamininer er nukleære intermediære filamenter som sammen med lamin-assosierte proteiner opprettholder kjernens struktur. Virker som støtte for kromosomer og replikerende DNA). Noen har leptin-mangel. Sekundære former til autoimmune sykdommer, metabolsk sykdom eller infeksjon (HIV).
Symptomer: Kongenitale former mistenkes ved påfallende mangel på subkutant fettvev, progressiv tap av fettvev på ekstremiteter samtidig med akkumulering av fett i lever og andre organer. Økt forekomst sammen med andre sykdommer (Garg A, 2011). Barn med manglende vektøkning, prominente muskler, vener, acanthos nigrans, xantomer, cushingoid eller akromegali-lignende utseende (Brown RJ, 2016).
- Familiær partiell lipodystrofi (FPL) sees ofte sammen med Emery-Dreyfuss muskeldystrofi (samme type mutasjon).
- Ved juvenil Dermatomyositt (JDM) har 25 % lipodystrofi. Sees også ved enkelte autoinflammatoriske sykdommer (JMP syndromet og Nakajo-Nishimuras sykdom)
- Andre relaterte tilstander
- Ehlers-Danlos syndrom
- Acrodermatitis chronica atrophicans (Borreliose)
- Hudtumorer
- HIV
- Diabetes (med særlig stort Insulin-behov)
- Alvorlig hypertriglyceridemi
- Fettlever (non-alkoholisk)
- Polycystisk ovarie syndrom (PCOS)
Vevsprøve: Lysmikroskopisk sees fiber atrofi og økning av intracytoplasmatisk fett.
Differensialdiagnoser: Alvorlig vekttap ved anorexia nervosa, sult, feilernæring, ukontrollert diabetes, hypertyreose, Addisons sykdom, cancer kakesi, alvorlig kronisk infeksjon. Donohue syndrom (leprechaunisme), Rabson–Mendenhall syndrom, multiple symmetrisk lipomatose. Cushing syndrom.
Behandling: Det finnes ennå ingen kurativ behandling. Assosierte autoimmune sykdommer og metabolske komorbiditer (diabetes, hyperlipidemi) må behandles optimalt. I noen tilfeller er leptin (metreleptin, Myalept®) indisert
Prognose: Alvorlig progressiv sykdom, men med ulike sykdomsforløp.
Litteratur:
Lysosomale lagringssykdommer/Avleiringssykdommer
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
Definisjon: Lysosomale lagringssykdommer omfatter ca. 50 diagnoser, mange av dem er svært sjeldne.
- Vennligst les om lysosomale lagringssykdommer i eget kapittel og om muskelmanifestasjoner i kapitelet om noen-inflammatorisk myopati.
Metabolsk syndrom
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
Metabols syndrom er en samling av flere tilstander som til sammen øker den individuelle risikoen for aterosklerotisk kardiovaskulær sykdom, insulin resistens, diabetes mellitus.
Årsakene er ofte multiple som overvekt eller fedme, mangel på fysisk aktivitet og genetisk disposisjon.
Litteratur: Swarup S, 2024
Mitokondriesykdommer
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
Mitokondrie-sykdommer (Kearns-Sayre syndrom med flere) er omtalt i kapitlet om myopatier
Mukolipidoser
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst se eget kapittel om lysosomale lagringssykdommer
Mukopolysakkaridoser
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst se eget kapittel om lysosomale lagringssykdommer
Osteoporose, osteopeni
Læringsmål REV 165. Revmatologen skal ha kunnskap om utredning og behandling av osteoporose.
Læringsmål REV 167. Revmatologen skal ha kunnskap om undersøkelsesprinsippene og beherske tolking av beintetthetsmåling, samt annen utredning og behandling av osteoporose.
Porfyri
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst se eget kapittel om porfyri
Purinmetabolisme
Den vanligste årsaken til urinsyregikt er vanligvis nedbryting av purin fra nukleinsyrer eller en redusert utskillelse gjennom nyrene. Sjeldne tilfeller er arvelig forstyrrelse av purinstoffskiftet (sjeldent) som Lesch-Nyhan syndrom. Allerede små barn produserer store mengder urinsyre og urinsyrekrystaller. Også nevrologiske symptomer kan oppstå.
- Vennligst se også Lesch-Nyhan syndrom ovenfor i dette kapitlet.
Skjelettsykdommer
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Osteonekrose, skleroserende skjelettsykdommer, kondrodysplasier (hos barn)
- Osteoporose og osteomalasi
- Paget sykdom
- Skjelettsmerter (differensialdiagnoser)
- Tumorer i ledd og leddnære strukturer og osteopetrose.
- Vennligst se også “Beinsykdommer” ovenfor i dette kapitlet.
Syre-base sykdommer
Læringsmål REV 107. revmatologen skal ha kunnskap om og vurderingskompetanse ved ulike syre-/baseforstyrrelser, herunder: respiratorisk acidose, respiratorisk alkalose, metabolsk acidose, metabolsk alkalose, «blandede typer».
Læringsmål REV 136. revmatologen skal ha kunnskap om de metabolske komplikasjonene ved nyresykdom, herunder elektrolyttforstyrrelser, syre-/baseforstyrrelser og forstyrrelser i mineralmetabolismen
Respiratorisk acidose skyldes respirasjonssvikt og akkumulering av karbondioksid (PCO2). PH i blodet synker. Nyrene forsøker å kompensere ved å utskille mer syre i form av hydrogen og ammonium, samt reabsorbere mer bikarbonat (Patel S, 2023). Supplerende informasjon kan leses i Metodebok i Indremedisin OUS-Ullevål (Akuttmedisin).
Metabolsk acidose skyldes økt hydrogenion-konsentrasjon (H+) i blodet, noe som medfører fall i bikarbonat (HCO3) til under 24 mEq/L. Årsaker er økt syreproduksjon (diabetes ketoacidose), redusert utskillelse av syre (nyresvikt), inntak av syre (acetylsalisylsyre) eller tap av bikarbonat (diare). Blodprøver måler nyrefunksjon, elektrolytter, venøs eller arteriell blodgass og toksiner ved behov. Tilstanden er potensielt alvorlig og viktig å korrigere. Behandlingen retter seg etter årsaken (Burger M, 2023). Supplerende informasjon kan leses i Metodebok i Indremedisin OUS-Ullevål (Akuttmedisin).
Alkalose skyldes overskudd av baser, slik at PH er >7,45. Multiple faktorer kan bidra til tap av hydrogen-ioner (H+) og overskudd av bikarbonationer (OH-). Alkalose kan være respiratorisk eller metabolsk, hvorav metabolsk alkalose er vanligst. Generelt er alkalose mindre alvorlig enn acidose.
-Respiratorisk alkalose skyldes høyt pulmonal CO2 tap ved hyperventilasjon (psykisk eller iatrogen eller initialt ved salisylat-overdosering som stimulerer respirasjonssenteret). For av CO2 produksjon kan ses ved koma under respiratorbehandling. Supplerende informasjon kan leses i Metodebok i Indremedisin OUS-Ullevål (Akuttmedisin).
-Metabolsk alkalose kan skyldes syre/H+ tap ved oppkast, økt bikarbonatinntak (enteralt ved melk-alkali-syndrom) eller parenteralt inntak av citrat eller acetat. Økt reabsorpsjon av bikarbonat via nyrene skjer ved alvorlig hypokaliemi, primær hyperaldosetronisme, Cushing syndrom, Bartter syndrom, Gitelman syndrom eller lakris-intoksikasjon. Også diuretika kan medføre økt bikarbonat absorpsjon. Supplerende informasjon kan leses i Metodebok i Indremedisin OUS-Ullevål (Akuttmedisin).
Thyreoidea sykdommer
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
- Vennligst se eget kapittel om thyreoidea-sykdommer
Urinsyregikt
- Vennligst se eget kapittel om urinsyregikt og avsnittet om “Purinmetabolisme” ovenfor.
Wilsons sykdom
REV 073. Revmatologen skal ha god kunnskap om aktuell behandling av metabolske tilstander assosiert med revmatisk sykdom, herunder hemokromatose og andre beinsykdommer.
Abnormal koppermetabolisme, ofte ved defekt ceruloplasmin produksjon. Osteoartritt (aggressiv artrose) hos 50% i håndledd. Det påvises periartikulær kalsifikasjon. Serum ceruloplasmin er ofte redusert (Chaudhry HS, 2023).
