BARN MED REVMATISK SYKDOM. BARNEREVMATOLOGI (REV 053-062)
125 Juvenil systemisk sklerose, JSSC, sklerodermi hos barn, lineær sklerodermi, morfea (REV 053, REV 054)
Øyvind Palm
Diagnosekoder: ICD-10: M34.8 Systemisk sklerose (andre former), L94.1 (lineær sklerodermi), L94.0 Lokalisert sklerodermi
Definisjon
Juvenil systemisk sklerose er den tredje vanligste revmatiske sykdommen i barnealder etter juvenil artritt og juvenil systemisk lupus. Den kan deles inn i to hovedgrupper (Foeldvari I, 2018):
- Systemisk sklerose Den mest alvorlige formen, som kan deles videre inn i diffus kutan form, begrenset kutan form og overlapp mellom disse.
- Lokalisert sklerodermi/morfea: Den vanligste formen.
Tidlig diagnostisering og behandling er viktig for en god prognose, og det er aktuelt å samarbeide med ulike pediatriske subspesialiteter og hudleger i oppfølgingen av pasientene. Lokalisert sklerodermi/morfea følges vanligvis opp av hudleger (Zulian F, 2018).
1. Juvenil systemisk sklerose: En multiorgansykdom
Juvenil systemisk sklerose er en multiorgan sykdom hos barn som medfører hard og stram hud, i tillegg til at andre organer kan angripes. Sykdommen skiller seg likevel fra systemisk sklerose hos voksne ved at indre organer angripes noe sjeldnere (se tabell nedenfor). Derimot kan forekomsten av samtidig artritt og myositt/dermatomyositt være noe vanligere (Stevens AM, 2019). Det anbefales at pasientene henvises avdeling med spesialkompetanse innen pediatrisk revmatologi (Foeldevari I, 2021).
Epidemiologi
Årlig insidens er i Storbritannia funnet å være 0,27-0,50 per million barn under 16 år (Herric AI, 2010; Pelkonen PM, 1994) og med en prevalens på 3 per. million (Beukelmann T, 2018). Sykdommen utgjør ca. 3% av systemisk sklerose når man også regner med den adulte formen. Jenter og gutter angripes like ofte før 8 års alder, deretter debuterer sykdommen tre ganger oftere blant jenter. Familiær forekomst er veldig sjelden (Mayes MD, 2003).
Symptomer
Sykdommen begynner aldri akutt, men utvikler seg over uker til måneder. Isolert Raynauds fenomen er typisk første symptom, etterfulgt av ødematøse fingre. Måneder til år senere kan pasientene oppleve stram hud, særlig på fingre og i ansiktet, teleangiaktasier og symptomer relatert til andre organmanifestasjoner (Stevens AM, 2019).
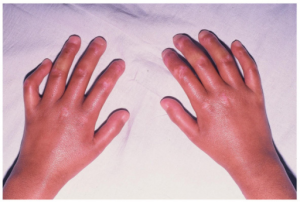
Hud: Vanligste debutsymptom er Raynauds fenomen. Flere måneder senere ses hudforandringer som kan omfatte:
- Hovne («puffy») fingre
- Hoven og stram hud særlig på fingre (sklerodaktyli) og i ansikt.
- Kalsinose ses hos 20-25%, oftest over albuer, MCP-ledd og knær.
- Teleangiektasier er sjeldnere.
- Ulcerasjoner, oftest på fingre.
- I ansiktet kan glatt, litt hoven og uttrykksløs hud være symptomer som leder en mot diagnosen (Zulian F, 2018).
Leddsmerter og artritt er ikke uvanlig og kan ses i tidlig sykdomsfase hos en av tre pasienter (Martini G, 2006).
Myositt er også vanligere enn blant voksne. Til sammen angripes 30% av barna av artritt og/eller myositt som kan forutgå sklerodermasymptomene (Scalapino K, 2006).
Gastrointestinale symptomer Øsofagusdysfunksjon kan påvises hos en av tre, men mange er asymptomatiske. Malabsorpsjon, diare og alvorlig vekttap kan ses ved langvarig sykdomsforløp (Martini G, 2006).
Kardiale symptomer: Fibrose kan medføre arytmier, og perikardvæske og kardial svikt forekommer også. Kardiale manifestasjoner kan påvises initialt hos 8,4 %, og senere i forløpet hos 24 %. Alvorlig kardial sykdom er ansvarlig for noen fatale forløp (Martini G, 2009).
Pulmonalt kan tørrhoste og belastningsdyspne indikere lungemanifestasjon. Pulmonal hypertensjon er en sjelden komplikasjon i barnealder (Martini G, 2006).
Nyreaffeksjon oppstår hos færre enn 5%, og da oftest som asymptomatisk proteinuri eller økt serumkreatinin som tegn på nyresvikt. Renal krise er uvanlig, men pasienter med anti-RNA polymerase III antistoffer og/eller topoisomerase I / Scl-70, samt de som får kortikosteroider er utsatt og må følges tett med blodtrykkskontroller og nyrefunksjon (Martini G, 2006).
Tabell. Kliniske forskjeller mellom juvenil og voksen/adult debut av systemisk sklerose (Stevens AM, 2019)
| Juvenil systemisk sklerose | Adult-onset Systemisk sklerose | |
|---|---|---|
| % (kumulativt i hele forløpet) | % (kumulativt i hele forløpet) | |
| Hud | 66–86 | 64–96 |
| Sklerodaktyli | 46–84 | 52–90 |
| Induration i hud | 64–86 | 77–96 |
| Perifert vaskulært | 84–100 | 91–100 |
| Raynauds fenomen | 84–100 | 91–100 |
| Digitale ulcera | 29–50 | 22–41 |
| Lunger | 36–55 | 44–64 |
| Pulmonal arteriell hypertensjon | 2–13 | 14 |
| Pulmonal fibrose | 9–26 | 22–36 |
| Kardiovaskulært | 5–18 | 11–25 |
| Hypertensjon | 3–8 | 11–17 |
| Kardial patologi | 2–17 | 3–20 |
| Muskelskjelett | 31–42 | 48–71 |
| Muskel svakhet | 20–32 | 15–27 |
| Artritt | 10–35 | 15–17 |
| Leddkontrakturer | 30–45 | 32–42 |
| Tendon friction rubs | 8–11 | 12–23 |
| Gastrointestinalt | 42–74 | 56–78 |
| Øsofagus | 24–60 | 65 |
| Mage | 16–30 | 26 |
| Ileum-kolon | 10–15 | 10–22 |
| Nyrer | 3–5 | 2–13 |
| Proteinuri | 3–5 | 6 |
| Renal krise | 0–4 | 2–13 |
Undersøkelser ved juvenil systemisk sklerose
Anamnesen. En bør kartlegge tegn til Raynauds fenomen (med ev. tid for debut), hovne fingre, sår på fingerpulpa, stramhet i huden på hender, ansikt og ellers på kroppen, samt svelgevansker, magesyre-oppstøt/refluks, fordøyelsesbesvær, vekttap og tegn til dyspne eller tegn til perifere ødemer. Spør også om tørrhetsplager fra øye eller munn (sekundært Sjøgrens syndrom). Følelse av hovne fingre og stramhet i ansikt er vanlige symptomer etter hvert.
Klinisk undersøkelse kartlegger aktuelle symptomer og mulige manifestasjoner i hud på fingre, tær, ansikt og truncus, samt indre organer. “Rodnan skin score” (se systemisk sklerose hos voksne) kan brukes til å estimere utbredelsen og omfanget av manifestasjonene.
-Initialt gjøres også en generell statusundersøkelse, som kan omfatte måling av blodtrykk, puls og vekt, samt auskultasjon av hjerte og lunger og palpasjon av abdomen. Huden på hender inspiseres og palperes for hevelse/«puffy fingers», sklerodaktyli, Ulcera/sår eller substansdefekter på fingertuppene, kalsinose og teleangiektasier. Kontrakturer beskrives. Ekstremiteter undersøkes for hudmanifestasjoner og bevegelighet.
Kapillaroskopi av neglefolder viser vanligvis tydelig patrologi allerede tidlig i forløpet. Utvidede kapillærer (megakapillærer), dreide kapillarer og blødninger er vanlige patologiske funn (Ingegnoli F, 2015).
Blodtrykk og puls (regelmessig/arytmi) og systematisk kartlegging av hjerte, lunger, abdomen gjøres.
Blodprøver omfatter CRP, SR, Hb, leukocytter, trombocytter, lever-, nyre- og thyreoidea-funksjonsprøver, elektrolytter, ANA med eventuelle subgrupper, inklusiv skleroderma-spesifikke antistoff (centromer/CENP, topoisomerase- 1 (Scl 70), fibrillarin, PM-Scl 70/100, RNA-polymerase I eller III). Ved artritt suppleres med anti-CCP. Ved mistanke om kardial manifestasjon kan NT-Pro-BNP være nyttig markør. Urin-stiks suppleres med kvantitering av protein og mikroskopi ved proteinuri.
Kardialt. EKG gjøres for å vurdere om arytmi foreligger. Ekkokardiografi brukes for å undersøke for ev. pulmonal hypertensjon eller økt perikardvæske. Ved mistanke om myokardfibrose (kardial svikt) kan MR med kontrast (late enhancement) gjøres.
- Lungefunksjonen kan testes blant eldre barn.
- CT eller HRCT av lunger er generelt en mer spesifikk undersøkelse for å påvise relatert interstitiell lungesykdom som kan være inflammatorisk eller fibrøs.
Gastrointestinalt kan øsofagus-dysfunksjon vurderes ved røntgen med kontrast-svelging eller manometri.
Kriterier for klassifikasjon
| PRES/ACR/EULAR her utarbeidet foreløpige klassifikasjonskriterier som krever minst ett major og to minor kriterier for diagnosen. Sensitivitet 90%, spesifisitet 96% (Zulian F, 2007). | |
| ORGANSYSTEMER
MAJOR kriterium |
KRITERIER
Sklerose/indurasjon i huden proksimalt for MCP leer MTP-ledd |
| MINOR kriteria | |
| -Hud | Sklerodaktyli |
| –Vaskulært | Raynauds fenomen |
| –Patologisk kapillaroskopi | |
| -Gastrointestinalt | Dysfagi |
| – | Refluks |
| -Renalt | Renal krise |
| – | Ny hypertensjon |
| -Kardialt | Arytmi |
| – | Hjertesvikt |
| -Respiratorisk | Pulmonal fibrose (CT lunger eller røntgen) |
| – | DLCO nedsatt |
| – | Pulmonal hypertensjon |
| -Muskel-skjelett | Tendon friction rub (senekrepitasjoner) |
| – | Artritt |
| – | Myositt |
| -Nevrologisk | Nevropati |
| -Serologi | ANA (Systemisk sklerose spesifikke antistoff (centromer/CENP, topoisomerase- 1 (Scl 70), fibrillarin, PM-Scl 70/100, RNA-polymerase I eller III) |
Behandling
Raynauds fenomen og digitale ulcera/sår på fingre kan behandles symptomatisk med varmehjelpemidler som hansker med varmetråder. Medikamentelt kan kalsiumblokkere, oftest nifedipin, forsøkes (OBS! hypotoni). Ved digitale ulcera eller behandlingsrefraktært alvorlig Raynauds fenomen er fosfodiesterase type 5 (PDE-5)-hemmere som sildenafil aktuelle. Alternativt kan bosentan brukes mot fingerulcera (Kowal-Bielecka O, Fransen J, 2017).
Sklerodaktyli og artritt. Ved progredierende, betydelig sklerodaktyli benyttes metotreksat, særlig ved samtidig artritt. Mykofenolat (CellCept) kan også ha effekt på slike hudmanifestasjoner, men neppe på artritt.
Lungemanifestasjoner. Ved alvorlig lungemanifestasjoner er mykofenolat aktuelt (Taskin DP, 2016), alternativt cyklofosfamid (Sendoxan) intravenøst månedlig i opptil 6 måneder eller utprøvende behandling med rituksimab. Hematopoetisk autolog stammcelle transplantasjon HMAS benyttes sjelden blant barn.
Gastrointestinale symptomer. Mot magesyre (refluks) benyttes protonpumpehemmere (PPI) som omeprazol eller lansoprazol. Ved alvorlig malabsorpsjon og bakteriell overvekt i tarm kan alternerende antibiotikabehandling med metonidazol, ciprofloksasin og doksycyklin være aktuelt i noen tilfeller.
Hypertensjon og nyreaffeksjon. Mot hypertensjon er ACE-hemmere (angiotensin converting enzyme-hemmere) aktuelle, også ved hypertensiv renal krise. Kortikosteroider som prednisolon skal brukes med forsiktighet fordi de kan utløse renal hypertensiv krise ved systemisk sklerose. Dersom myositt eller uttalt artritt preger sykdomsbildet, kan likevel prednisolon i lave doser være indisert.
Prognose
Indre organer angripes sjeldnere enn hos voksne med systemisk sklerose, og prognosen er derfor generelt bedre. En langsom progresjon av kutane manifestasjoner må likevel forventes. Ved overlappsformer kan graden av myositt og av artritt være av betydning. Hjerte- og lunge-komplikasjoner er generelt de mest alvorlige.
Oppfølging
Det anbefales at alle barn med systemisk sklerose følges minst hver 6. måned med kontroll av lungefunksjon (inkludert DLCO), kardial ekkokardiografi, nyrefunksjon. Huden bør også vurderes regelmessig ved hjelp av Rodnan hudskår (Foeldevari I, 2021).
2. Lokalisert sklerodermi
")
Definisjon
Lokalisert sklerodermi er en sykdom begrenset til hud og underhud, eventuelt også underliggende muskler, skjelett, nerver og annet vev. Indre organer som hjerte, lunger, nyrer og tarm angripes ikke. Sykdomsårsaken er ukjent, men dysregulering av fibroblaster og produksjon av kollagen spiller en sentral rolle i patogenesen. Komorbiditet med klassiske autoimmune sykdommer som autoimmun thyreoiditt, vitiligo og diabetes mellitus type 1 indikerer en autoimmun årsak (Leitenberger IJ, 2009).
Epidemiologi
I Storbritannia er den årlige insidensen 2,5-3,4 per million barn. Prevalensen er beregnet til en per 100.000 (Beukelmann T, 2019). Denne begrensede formen er dermed omtrent ti ganger vanligere enn den systemisk sklerose blant barn. De fleste har den lineære typen (se nedenfor). Tilstanden er noe vanligere blant jenter enn gutter (ratio 2,4:1), og gjennomsnittlig alder ved sykdomsdebut er 7,3 år (Herrick AL, 2010). Det finnes også en medfødt/kongenital form (Zulian F, 2006).
Klassifikasjon
Lokalisert sklerodermi kan deles inn i fem undertyper (Zulian F, 2007);
- Cirkumskript morfea er begrensede ovale eller runde indurerte, gulhvite områder med fiolett halo, oftest på truncus, sjeldnere på ekstremiteter eller ansikt.
- Generalisert morfea består av fire eller flere lesjoner som er over 3 cm i diameter og fordelt på minst tre av syv anatomiske områder (hode-hals, høyre arm, venstre arm, høyre ben, venstre ben, anteriore truncus og posteriore truncus).
- Lineær skleroderma er vanligste subtype blant barn og ungdom. Typisk ses en lineær, atrofisk strek i huden over panne, hodeskalle (“en coup de sabre”), ekstremitet og/eller truncus. Subkutan hud, fettvev og andre underliggende strukturer angripes ofte, noe som kan føre til redusert lokal vekst og skjevheter. Parry-Romberg syndrom er en variant der ansiktet kaudalt for pannen er angrepet, noe som lett medfører asymmetri og problemer med kjevefunksjonen (Arif T, 2020; E-Kehdy J, 2012).
- Pansklerotisk morfea er svært sjelden, men alvorlig fordi så store deler av huden angripes, slik som truncus, ekstremiteter, ansikt og hodebunn. Til forskjell fra systemisk sklerose spares hender og føtter. Raynauds fenomen er heller ikke vanlig. I forløpet kan likevel dype strukturer som blodårer skades, slik at blodsirkulasjonen kan bli alvorlig redusert.
- Blandet subtype av morfea har innslag av to eller flere av de ovenfor nevnte subgruppene.
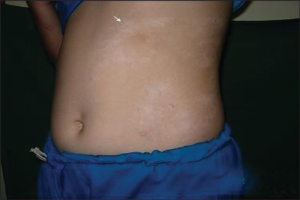
Ekstrakutane manifestasjoner ved lokalisert sklerodermi
Selv om lokalisert sklerodermi/morfea primært rammer hud og underhud, kan det også oppstå manifestasjoner utenfor huden. Artritt forekommer hos 20% av pasientene, ofte i andre områder enn der huden er affisert. Nevrologiske manifestasjoner ses særlig ved lineær sklerodermi, i form av hodepine, kramper og læringsvansker. MR-underøkelse av hjernen kan vise lesjoner i hvit substans, vaskulære malformasjoner og kalsinose.
Undersøkelser
Anamnesen fokuserer på sykdomsdebut og utbredelse over tid. Raynauds fenomen og andre symptomer fra hender og føtter er uvanlige. Spør om symptomer fra ledd (artralgi, artritt) og nervesystemet .
Klinisk undersøkelse vurderer utbredelsen i huden i henhold til klassifikasjonen nevnt ovenfor, ledd- og muskel-symptomer, samt nevrologiske kjennetegn.
Laboratorieprøver kan omfatte CRP, SR, Hb, leukocytter med differensialtellinger, trombocytter, elektrolytter, lever-, nyre og thyreoidea-funksjonsprøver, IgG, samt ANA og anti-CCP. Urin stiks. Forhøyet IgG og/eller ANA forekommer. Blant ANA-subgrupper er anti-histon antistoff, særlig ved generalisert morfea. Antifosfolipid antistoff (lupus antikoagulant, kardiolipin- og beta-2 GP) er uvanlig.
Bildediagnostikk: MR av angrepet hud viser hvor dypt lesjonen går og om annet vev er angrepet. MR av hjernen er viktig dersom ansikt eller skalpen er angrepet, for å kartlegge eventuelle forandringer i hjernen, øyne og kjeve. Andre metoder som infrarød termografi, laser Doppler flowmetri og høyfrekvent ultralyd kan også benyttes, men disse har varierende spesifisitet og tolkning av resultater er avhengig av undersøkerens erfaring.
Behandling av lineær sklerodermi
Lokaliserte, benigne, langsomt progredierende former behandles med lokale steroider. Mer aktive, økende forandringer bør vurderes for systemisk behandling der metotreksat oftest brukes. Prednisolon i en kort periode for metotreksat virker kan også være aktuelt. Mykofenolat er et alternativ til metotreksat. Utprøvende behandling med takrolimus og biologiske legemidler (TNF-hemmere eller IL6 hemmere) er prøvd i behandlingsrefraktære tilfeller.
Prognose
Lokalisert sklerodermi har generelt en god prognose. Typisk er en tidlig inflammatorisk fase med senere stabilisering. Gradvis spontant mykere hud er deretter ikke uvanlig, men pigmentforandringer kan bestå. En studie viste at 3,8 år etter diagnosen hadde 50% mykere hud (Peterson LS, 1997). Likevel vil over 50% med lineær sklerodermi ha vedvarende redusert funksjon på grunn av sykdommen (Piram M, 2013).
Differensialdiagnoser
- Borreliose (sen manifestasjon av hud-manifestasjon)
- Darier’s disease (DAR)
- Dermatomyositt (Juvenil)
- Eosinofil fasciitt
- Fenylketonuri
- Graft-versus-host sykdom (kronisk GVHD )
- Juvenil artritt
- Lipodystrofi
- MCTD
- Melorheostose
- Mutasjon i MAP2K1 gen
- Nefrogen systemisk fibrose (NSF)
- Overlapp syndrom med innslag av flere bindevevssykdommer
- Palmoplantar keratoderma/skleroatrofisk syndrom/Huriez syndrom
- Pannikulitt
- Prematur aldring syndromer (inklusiv Progeria og Werner syndrom)
- Sarkoidose i huden
- Systemisk Lupus (SLE)
Retningslinjer
SHARE; Foeldevari I, 2021 (management)
Nasjonale retningslinjer for lokalisert sklerodermi (fra NAKBUR)
Litteratur
Foeldevari I, 2021 (management, SHARE konsensus)
Zulian F, 2007 (Juvenil systemisk sklerose)
