BINDEVEVSSYKDOMMER (REV 021-033)
63 Systemisk sklerose (SSc). Sklerodermi (REV 021)
Jan Tore Gran and Øyvind Palm
Kjennetegn på systemisk sklerose
Raynauds fenomen med debut i voksen alder.
Hovne/”puffy” fingre ved debut, stramhet i huden, særlig på fingre (sklerodaktyli).
Antistoff hos> 50%: CENP, Scl-70 og RNP-polymerase III er karakteristiske, men foreligger ikke hos alle.
Alvorlige organmanifestasjoner omfatter skleroderma renal krise, pulmonal hypertensjon, lunge-fibrose og GAVE (gastric antrum vascular ectasia).
Diffus kutan systemisk form og begrenset form har ofte ulike forløp
Diagnosekoder ICD-10: M34.0 (diffus form), M34.1 (begrenset form/CREST). (J99.1*) Lungeaffeksjon† ; M34.9 Uspesifisert systemisk sklerose
Prosedyrekoder: 6-minutter gangtest: FYFX05. Kapillærmikroskopi: PKFT00. EKG: FPFE15
ATC koder (for legemiddelstatistikk): L04A A Immunsuppressive legemidler:
Definisjon
Systemisk sklerose (Ssc), også kalt systemisk sklerodermi, er en sjelden autoimmun bindevevssykdom. Den kjennetegnes ved progredierende fibrose i hud og underhud. Sykdommen rammer både kvinner og menn i alle aldre, og kan være mild eller alvorlig. Ssc kan deles inn i tre hovedtyper:
- Begrenset kutan form (begrenset Ssc). Denne typen rammer kun huden på hender, føtter og omkring munnen. Den er ofte assosiert med CENP-antistoffer.
- Diffus Ssc: Denne typen rammer huden på hele kroppen og kan også involvere indre organer. Den er ofte assosiert med Anti-Scl70-antistoffer (Volkmann ER, 2023).
- Dersom sykdommen den begynner hos barn, kalles den juvenil systemisk sklerose.
Systemisk sklerose skilles fra non-systemisk sklerodermi (morfea, lineær sklerodermi) som angriper huden, men ikke indre organer.
Historikk
Legen Carlo Curzio fra Napoli beskrev i 1753 en 17 år gammel pike (Patrizia Galiera) med en tilstand mest sannsynlig representerte SSc. Maurice Raynaud i 1863 og Jonathan Hutchinson i 1883 påpekte sammenhengen mellom Raynauds fenomen og skleroderma. Gintrac foreslo betegnelsen sclerodermie i 1847. Den første hudbiopsien beskrev Kohn (Kaposi) i 1869. Han konkluderte med at det forelå fortykket lymfe i hudens bindevev (Kaposi M. Pathologie und Therapie der Hautkrankheiten. 4th ed. Vienna, Austria: Urban & Schwarzenberg, 1893). CREST syndromet først beskrevet i 1910 (Thiberge-Weissenbachs syndrom). I 1957 beskrev en gruppe fra Mayoklinikken i USA ulike typer sklerodermi, inklusiv systemisk sklerose. De påpekte funn av fibrosis og skeroserende kollagenfibre, samt, negative tester for mucin (O’leary P, 1957). I 1979 publiserte Rodnan en skåringsmetode for manifestasjon i huden (Rodnan GP, 1979). Metoden som fortsatt er mye brukt er en “modifisert Rodnan skin score” (Brennan P, 1992). Forekomsten (prevalens) i Norge ble estimert i 2012, basert på en befolkningsbasert studie i Sør-Øst Norge (Hoffmann-vold A-M, 2012).
Epidemiologi
Systemisk sklerose angriper fire ganger så mange kvinner som menn og vanligste debutalder er i 30-60 årene. Den forekommer også blant barn (Juvenil systemisk sklerose), men meget sjelden hos menn under 30 års alder. Prevalens i sør-øst Norge er beregnet til 9,9/100.000 (begrenset form 6,9/100.000, diffus form 1,8/100.000) med en gjennomsnittsalder ved debut på 47 år. Den diffuse formen begynner oftest blant relativt unge, mens den begrensede formen er vanligere i litt høyere alder. Gjennomsnittsalder ved sykdomsdebut er 47 år (Hoffmann-vold A-M, 2012).
Etiologi
Genetiske disponerende faktorer er undersøkt først i tvillingstudier og senere i store, multisenter hel-genom/genome-wide association studies (GWAS) studier. Disse har identifisert mange gener som er involvert i kontroll av inflammasjon og inflammasjon og antakelig også relasjoner til sykdomsdisposisjon (Feghali-Bostwick C, 2003; Broen JCA, 2014).
Miljøfaktorer som kan medføre sklerodermi-lignende sykdom er påvist. Disse omfatter silikonstøv, medikamenter og forurensede matvarer.
Selv om den eksakte mekanismen ikke er vist, er det sannsynlig at en kombinasjon av genetisk disposisjon og uheldig påvirkning av miljøfaktorer utløser sykdommen (Rosendahl A-H, 2022).
Patogenese
De karakteristiske patofysiologiske forandringer ved systemisk sklerose kan ses allerede ved rutinemessig histologisk undersøkelse: Tidlig i forløpet ses ødematøse forandringer i endotelceller, etterfulgt av lymfohistiocytiske inflammatoriske infiltrater rundt affiserte blodkar. Senere dannes depoter av ekstracellulær matriks med aktiverte myofibroblaster og homogene kollagen bunter (Rosendahl A-H, 2022).
Tre abnormiteter kjennetegner SSc: 1) Fibroblast dysfunksjon som fører til økt deponering av ekstracellulær matriks. 2) Vaskulære forstyrrelser (vaskulopati) som gir vevshypoksi. 3) Immunrespons kjennetegnet ved endret T- og B-Iymfocytt-funksjon og produksjon av auto-antistoffer. Sannsynligvis er den initiale hendelsen en endotel-skade som affiserer mikrokar og små muskulære arterier. Dette medfører tap av kapillærer (destruktiv vaskulopati) som ikke kompenseres på grunn av dysfunksjon av både angiogenese og vaskulogenese.
Angiogenese er dannelse av nye kar ved såkalt “sprouting”-dannelse med utgangs-punkt i allerede eksisterende kar.
Vaskulogenese er dannelse av nye kar ved sirkulerende progenitor-celler, altså uavhengig av allerede eksisterende kar. Imidlertid er de fleste proangiogenetiske faktorene oppregulert ved SSc (VEGF, vascular endothelial growth factor), men kan altså ikke oppveie økningen i angiostatiske faktorer. En alternativ teori er at kapillærtapet ikke skyldes redusert mengde dannelse av nye kar, men feil i modningen av disse. Hva som forårsaker den initiale endotelskaden er ukjent, men både infeksjoner, oksidativt stress, hypoksi, hyperlipidemi og inflammasjon trigger endotelceller. Hvilken rolle autoantistoffer som topoisomerase-1 (Slc-70) og anticentromer antistoffer (CENP) spiller for den tidlige inflammasjonen og påfølgende endotelskade, er ukjent.

Endotelskaden medfører også vaskulær remodellering med hypertrofi av intima og media, samt fibrose av adventitia. Disse forandringene fører til tiltakende forsnevring av karlumen og obliterasjon (proliferativ vaskulopati). Den proliferative vaskulopatien medieres av en rekke molekyler som for eksempel økt produksjon av endotelin, redusert NO syntetase-produksjon og nedsatt frigjøring av prostacycliner. Kapillær-tapet gir kliniske symptomer som akrocyanose og digitale ulcera, mens vaskulær remodellering gir pulmonal arteriell hypertensjon og skleroderma nyrekrise.
Økt fibrosering er selve kjennetegnet på SSc og består av en ukontrollert produksjon av kollagen og andre ekstra-cellulære matriks (ECM) proteiner av fibroblaster. Deponeringen av kollagene fibre skjer i den retikulære delen av dermis med avsmalning av den papillære delen. Tidlige hudforandringer er ødem, perivaskulære infiltrater og degenerering av kollagene fibre. De patologiske forandringene er imidlertid de samme for alle affiserte organer. Den akkumulerte mengden proteiner i ECM forstyrrer vevsarkitekturen og medfører dysfunksjon av dens funksjoner. De prolifererende fibroblastene finnes allerede i nærheten av endotelskaden, men i tillegg aktiveres og differensieres mesenkymale progenitor-celler, epitelceller og endotelceller til fibroblaster, hvilket forsterker den fibroserende prosessen. Fibroblastene transformeres så til myofibroblaster, som har kontraktile egenskaper, og er svært motstands-dyktige overfor apoptose. Det patologiske sluttresultatet er atrofi og hardhet (sklerose) av affisert vev (Kormann B, 2019).
Symptomer
Raynauds fenomen er det klassiske første symptomet på systemisk sklerose. Imidlertid er Raynauds fenomen veldig vanlig i befolkningen (8-10%) og således lite spesifikt. Typisk ved systemisk sklerose er at Raynauds fenomen oppstår i voksen alder og øker i intensitet. Ved begrenset form for systemisk sklerose er latenstid mellom Raynauds fenomen og andre symptomer gjennomsnittlig 5 år. Tilsvarende for diffus form er 1-2 år. Individuelle variasjoner er imidlertid betydelig (Walker UA, 2007; LeRoy EC, 1988). Initialt sees den klassiske triaden med avblekning, cyanose og reaktiv hyperemi. Med tiden består ofte anfallene bare av cyanose (“blue attacks”), og til slutt preges tilstanden av kronisk akrocyanose. Ved uttalte Raynauds fenomen kan fingre og tær få iskemiske, nekrotiske sår (ulcera, se nedenfor) med påfølgende substanstap distalt. Digitale ulcera kan utvikles tidlig i sykdomsforløpet.

Allmenn-symptomer som tretthet/fatigue og vekttap ledsager ofte. Artralgier og myalgier er vanlige. Ved begrenset form debuterer organmanifestasjonene oftest i senfasen (etter fem års sykdom). Samtidig preges sykdommen da ellers av stabile hudforandringer (fibrose) og lite uttalte allmennsymptomer. Ved diffus form ses organmanifestasjoner ofte tidligere sykdomsfase (særlig første to år dra sykdomsdebut).
Debut av organmanifestasjoner utenom Raynauds fenomen ble i EUSTAR-kohorten på 695 pasienter funnet å oppstå median 0,9 år etter Raynauds hos 87% av pasientene. Manifestasjonene utviklet seg deretter noe ulikt. Etter ett år og etter fem år var andel med manifestasjoner fra henholdsvis hud, lunge eller GI-trakt økt fra ca. 70% til 85%. Tilsvarende for digitale ulcera, lunge-påvirkning (FVC<80%) og for hjerte-manifestasjoner (diastolisk dysfunksjon, rytmeforstyrrelser, perikardvæske, redusert pumpefunksjon/LVEF <50%) fra ca. 30% til 50%, for pulmonal hypertensjon fra ca. 10% til 25% og for renal krise fra ca. 5% til 10%. Pasienter med anti-ScL70 (anti-topoisomerase) og diffus form var generelt mer utsatt enn ved CENP/anti-centromer og begrenset form. Anti-RNA polymerase III disponerer klart for renal krise (Jaeger VK, 2016).
Huden angripes hos nær 100 % av pasienter med systemisk sklerose. Sklerodermi sine skleroderma (se ovenfor) er et unntak, men de fleste av disse utvikler hudlesjoner etter hvert.
Hudforandringene starter vanligvis distalt på fingrene. Ødematøse forandringer (“puffy”) er tegn på typiske mikrovaskulære forandringer. Hudtykkelse og utbredelse kan registreres ved modifisert Rodnan hudscore (vennligst se Undersøkelser nedenfor). Huden blir fortykket og stram, ofte med sprekk-dannelser og digitale ulcera sentralt på fingerpulpa. Områder med de- eller hyperpigmentering (“salt og pepper”) iakttas hyppig in forløpet, særlig ved den diffuse formen. Ved begrenset form kan pasienten ha Raynauds fenomen i mange år før huden affiseres. Ved diffus form utvikles Raynauds uker til få år før, samtidig med eller like etter hudmanifestasjonene. Raynauds fenomen forekommer hos nesten alle pasienter. Klassisk Raynauds med avblekning, cyanose og reaktiv hyperemi ved temperaturfall. To av tre symptomer skal være til stede. I løpet av sykdommen inntreffer ofte en mer permanent kompromittering av mikrosirkulasjonen. Det kliniske bildet blir da mer lik akrocyanose.
. CC BY-NC-ND 4.0")
Puffy hands/fingers preges ved diffus form av raskt innsettende hudforandringer: ødem (“puffy hands”, dvs. diffus ødematøs hevelse). Ved begrenset form kan forandringene utvikle seg over flere år. Symptomene begynner gjerne distalt i hender for så å bre seg proksimalt. Vanligvis utvikles forandringene på flere fingre på begge hender omtrent samtidig. Huden føles litt stram og fingrene kan være vanskelige å strekke helt ut. “Bowed fingers” er et tidlig tegn. Årsaken er endotel dysfunksjon, slik som også ved Raynauds fenomen og kutane ulcerasjoner (Zania-Silva DC, 2021). En bør være oppmerksom på at puffy hands og Raynauds fenomen også er karakteristisk for MCTD.
-“Scleroderma neck sign” angir tilstedeværelsen av et stramt bånd over platysma ved hyperekstensjon av nakken. Forekommer ved diffus form og kan palperes hos over 90 %.
-Roman breastplate. Senere kan truncus affiseres (“Roman breastplate”) ved diffus form.
-Karpemunn utvikles ved affeksjon av munn og periorbalt vev, ofte ved begrenset form.
-Tendon friction rub palperes som “kram snø” og signaliserer alvorlig sykdom. Forekommer oftest ved aktiv, diffus kutan sykdom.
-Digitale ulcera er vevs-lesjoner som kan utvikles til nekrose og som involverer epidermis, dermis og subkutant vev. Ischemiske sår utvikles hos opptil 40 % av pasienter. De lokaliseres typisk ved benete fremspring (over PIP-ledd) og ved endearterier (fingerpulpa), men kan også sees proksimalt og distalt på underekstremitetene langt fra benete fremspring. Hyppigst på andre og tredje finger. Opp mot 11 % av slike ulcera ender med gangren eller amputasjon, og risikoen er størst for sår som varer lengre enn 6 måneder. Større iskemiske nekroser er uvanlig.
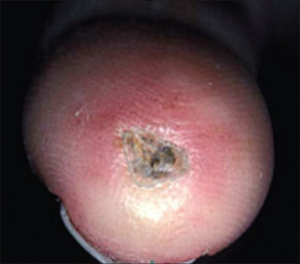
-Skorper og fissurer skyldes dermal fibrose på områder der huden er tynn og atrofisk. Små overfladiske ulcerasjoner (1-4 mm.) er oftest forårsaket av okklusjon av prekapillære arterioler. Dype sår på distale fingre og tær kommer som resultat av okklusjon av større kar eller sykdomsprosesser i kutane ekstremitetskar. Skarpt avgrensede iskemiske sår, ev. med tap av fingre eller tær, er også forårsaket av kar-okklusjon og ledsages gjerne av smerter (kritisk iskemi). Ved kritisk iskemi opptrer smerter, og proksimalt for det iskemiske området sees hyperemi.
-Pitting scars representerer vevstap og bør dermed ikke oppfattes som digitale ulcera. Lokaliseres til finger-pulpa og på lateralsiden av fingrene.
–Kalsinose ses hos ca. 25% av pasientene oftest ved begrenset form av systemisk sklerose. Vanlig lokalisering er på fingre, albuer og knær. Kalsinose ses også ved dermatomyositt og andre tilstander (Valenzuela A, 2018).
-Sår som ikke tilheler kan skyldes andre forhold enn grunnsykdommen. Det er viktig å utelukke kalsinose, ateromatose, infeksjon, diabetisk angiopati og koagulopati (inklusiv antifosfolipid syndrom). I sistnevnte gruppe bør man også utelukke Faktor V Leiden mangel som er den mest vanlige koagulopati. Biopsi vil her vise fibrinnedslag, men ingen inflammasjon. Venøse leggsår må også utelukkes. Disse gir spreng og tyngdefornemmelse i oppreist stilling, lokal kløe, parestesier og ikke sjelden nattlige kramper. Teleangiektasier, corona phlebectatica (vifte-formede teleangiektasier), brunlig hyperpigmentering og eksem er ikke uvanlig. Lipodermatosklerose kan utvikles etter noe tid (se også differensialdiagnoser nedenfor). Tegn til venøs insuffisiens med varicer sees ofte.
Sklerodaktyli. Med sklerodaktyli menes hard og noe tykk hud distalt for MCP-ledd i fingre. Begrenset form forutgås av Raynauds fenomen ofte med flere år. Mange med denne formen utvikler hud-forandringer begrenset til fingre og rundt munnen. Pre-skleroderma og tidlig systemisk sklerose er beskrevet ovenfor.
Pitting scars er substansdefekter på fingerpulpa. De oppstår spontant, ofte litt ut i sykdomsforløpet og er lokalisert ganske sentralt på fingertuppene. En må skille disse fra sår ved subkutan kalsinose som heller ikke er uvanlig ved sykdommen.
. CC BY-NC 3.0")
Lunge-manifestasjoner merkes oftest relativt sent i forløpet. Tørrhoste eller belastningsdyspne er vanligste tegn. Forandringer kan påvises hos 50-65% av pasientene og opptrer både som sykdom i lungevevet og vaskulær lungeskade. Førstnevnte benevnes oftest interstitiell lunge-sykdom (ILD), selv om parenkymet representert ved alveolene ofte er angrepet. Symptomene preges av dyspné og hoste. Den raskeste utviklingen av ILD skjer ofte de første 4 årene etter diagnosen (Hoffmann-Vold A-M, 2019).
-Pulmonal hypertensjon (PAH). Vaskulær sykdom i lungearterier medfører pulmonal arteriell hypertensjon (PAH) hos 8-12% av pasientene (Aithala R, 2017). Symptomer kan være dyspne og utmattelse, men er ofte asymptomatisk (Morrisroe K, 2017) og trenger ikke være ledsaget av annen lungesykdom. Fordi PAH er årsaken til død hos mer enn halvparten av pasientene med systemisk sklerose (Kolstad KD, 2018) og tidlig terapi er viktig, er PAH-screening sterkt anbefalt, også blant asymptomatiske pasienter (Gailie N, 2016). Med tidlig behandling menes at pasientene er i WHO funksjonsklasse I eller II av maksimalt IV mulige. Klasse II tilsier lett begrenset aktivitet. Ved regelmessig screening er 8-års overlevelse funnet å kunne økes fra 17% til 64% (Humbert M, 2011). Nedsatt DLCO (gass diffusjonskapasitet) ved lungefunksjonstester kan predikere utvikling av både ILD og PAH. DLCO eller karbon mono-oksid transfer faktor måler gassutvekslingen i nivå med den alveolære membranen og tilkjennegir dysfunksjon i overføringen av CO. Dysfunksjonen kan skyldes redusert alveolær overflate eller nedsatt membranfunksjon. En FVC/DLCO ratio > 1,4 peker mer i retning av PAH enn av ILD. Vennligst les mer om PAH i eget kapittel
-Interstitiell lungesykdom (ILD) i form av fibroserende alveolitt er en interstitiell, retikulær type lungesykdom som preges av fibrose og som er lite tilgjengelig for behandling. Utprøvende antifibrotisk medikasjon med nintedanib (Ofev) kan redusere sykdomsprogresjonen mer enn placebo, men den kliniske nytten er usikker (Distler O, 2019). I enkelte tilfeller er lungetransplantasjon aktuelt (Shah RJ, 2017). Den inflammatoriske typen ILD kan derimot respondere på immunsuppresjon. Imidlertid vil det ofte foreligge en blanding av fibrose og inflammasjon. Å avgjøre hvilke av de to prosessene som dominerer hos den enkelte pasient kan være avgjørende for valg av behandling. Histologisk er NSIP (non-specific interstitial pneumonia – homogene forandringer som affiserer alle lungepartier) den hyppigste typen ILD ved SSc, fulgt av UIP (usual interstitial pneumonia – deler av lungen affisert, ofte honeycombing). Vennligst les om NSIP og UIP i eget kapittel om lungemanifestasjoner ved revmatisk sykdom.
Nyresykdom med nyrekrise (scleroderma renal crisis, SRC)
Nyrekrise (“Scleroderma renal crisis” – SRC) sees nesten bare ved diffus type SSc (10-25%) og da oftest hos pasienter med anti-RNA polymerase III antistoffer (33% av dem med dette antistoffet). Symptomene er ny hodepine, utmattelse, svimmelhet, neseblødninger og andre symptomer som følger av hypertoni. Omtrent 75% debuterer i løpet av de første fire sykdomsårene (median 8 måneder), men er også beskrevet (sjelden) etter 20 års sykdom. Som regel foreligger ingen forutgående hypertensjon. SRC karakteriseres av akutt stigning av blodtrykket (> 160/90), retinopati > grad Ill og raskt avtagende nyrefunksjon. SRC kan ledsages av alveolær kapillaritt (lungeblødning) og er da et pulmonalt-renalt syndrom (Prabhakar N, 2023). -Patogenese ved renal krise. lnitialt foreligger en endotelskade som gir fortykkelse og proliferasjon av intra-lobulære og arcuate kar. Dette resulterer i plate-aggregasjon, økt kollagen og fibrindeponering som ytterligere kompromitterer karlumen. Resultat en nedsatt nyreperfusjon som gir hyperreninemi. Plasma-renin er forhøyet 10-100 x øvre normalverdi. Også serum Endothelin-1 stiger. -Risikofaktorer. Utenom anti-RNA polymerase III antistoffer ved diffus form som disponerende faktor er episoder er utløst av hjertesvikt (nedsatt nyreperfusjon), perikardvæske, arytmier, sepsis, dehydrering. Økt risiko ses også ved høydose prednisolon (> 10-15 mg/d) siste 6 måneder. Palpabel «tendon friction rub» øker også risiko for SRC. Bruk av ciclosporin kan utløse raskt innsettende nyresvikt som minner om SRC. Plasma-renin-nivå forut for nyresykdom sier intet om risiko for å utvikle SRC. -Klinisk ytrer nyrekrise seg ved alvorlig hodepine, synsforstyrrelse eller encefalopati-symptomer. Kramper, økt tretthet, kvalme, konfusjon sees også. Over 90 % har hypertensjon, 30 % har diastolisk trykk > 120 mmHg. Av pasienter med SRC er omkring 10 % normotensive. Disse kan allikevel ha trykkstigning, men innenfor det vi oppfatter som normalverdier. -Laboratorieprøver viser ofte redusert Hb med mikroangiopatisk, normokrom hemolytisk anemi der schistocytter er typisk i blodutstryk. Retikulocytose og trombocytopeni > 50 000) ses hos omkring halvparten. Andre blodprøveutslag kan være stigende LD, troponin, NTpro-BNP, mens haptoglobin, og C3/C4 kan falle. Proteinuri oftest < 2g/24t. Mikroskopisk hematuri og kornede sylindre. Serum-kreatinin stiger daglig, selv etter at BT er normalisert. -Nyrebiopsi. Systolisk blodtrykk >160 mmHg kan være kontraindikasjon for biopsi. Biopsi tas når BT og koagulasjonsstatus er normalisert (Penn H, Howie AJ, 2007; Chrasaszcz M, 2020). Histologisk ses infiltrasjon av glatt muskelceller og deponier av kollagen i intima som medfører konsentrisk fibrose, såkalt “løk-skall utseende”. -Behandling av hypertensiv nyrekrise er essensiell. Etter at ACE-hemmere ble tilgjengelige har ett års mortalitet falt fra 85% til 24% (Zanatta E, 2018). Likevel vil en del av pasientene få behov for nyretransplantasjon på sikt (Prabhakar N, 2023). Vennligst se mer om behandling i avsnitt senere i dette kapitlet.
Alvorlige kardiale komplikasjoner forekommer i form av myokarditt, hjertesvikt, perikarditt, cor pulmonale, asymptomatisk kardial fibrose og patologisk ventrikulær relaksasjon. MR-undersøkelser gjøres ikke rutinemessig, men har bidratt til å avdekke flere tilfeller (Bissel L-A, 2017).
-Pulmonal hypertensjon. Årlig ekkokardiografi anbefales for å oppdage tidlige tegn på pulmonal hypertensjon. Kardiolog bør oppgi mål på høyre atrium areal (cm2) og hastigheten på trikuspidal-klaff-tilbakestrøm (TR hastighet i m/s) som inngår i DETECT-kalkulatoren. Ved mistanke om pulmonal hypertensjon kan DETECT-kalkulator brukes for å estimere behov for høyre-kateter-hjerteundersøkelse. Kalkulatoren benytter data fra EKG, ekkokardiografi, hjertefrekvens, blodtrykk, NT-proBNP og urat-nivå (Young A, 2021).
–Myokardfibrose undersøkes ikke rutinemessig, men kan påvises ved MR-undersøkelse av hjertet hos 45% (Rodriguez-Reyna TS, 2015, Ramalho AR, 2017, Ntusi NA, 2014). En mistanke om myokardfibrose kan en få ved hjertesvikt, først i form av diastolisk dysfunksjon som kan påvises ved ultralyd Doppler av hjertet (Smiseth OA, 2019). Diagnosen sikres hvis myokardbiopsi utføres. Vennligst se også kapittel om Pulmonal hypertensjon. Hjertemanifestasjoner ved revmatiske sykdommer er også beskrevet i eget kapittel
Omtrent 90% av pasientene har en form for gastrointestinal manifestasjon (McMahan ZH, 2019).
-Øsofagus-dysmotilitet av distale 2/3 med tilhørende svelgeproblemer, samt dyspepsi og refluks er meget vanlig (> 80 %). Diagnosen sikres med røntgenkontrast, manometri eller scintigrafi. Hovedsymptomet ved gastro-øsofageal refluks er bryst-brann som oppleves som et oppadstigende, sviende ubehag fra epigastriet opp mot sternum. Symptomene inntreffer som oftest få timer etter måltid og kan forverres ved fremover-bøying eller ved horisontalt leie. Ved øsofagus betingede svelgevansker kan også pasientene ha følelse av klump i halsen og smerter ved svelging. Ved alvorlig refluks kan Barretts øsofagus utvikles (dysplastiske slimhinneforandringer), hvorav 10 % overgår i adenokarsinom.
–Dysfagi kan ha ulike årsaker. Ved forstyrrelse av den orale svelgfasen plages pasienten ofte av at maten samler seg i munn og gane, samt ved svakhet i tungen. Ved forstyrrelser i den faryngale fase beretter pasientene ofte om nasal regurgitering, hoste etter svelging og stadig gurglete stemme.
-Gastroparese med forsinket tømming gir anoreksi, metthetsfølelse, kvalme og evt. oppkast. Malabsorpsjon (mangel på vitamin B12, vitamin D, kalsium, jern og kalorier) og kolon-sakkulasjoner (innsnevringer) sees. Nedsatt motilitet kan gi pseudoobstruksjon og bakteriell overvekst.
-Watermelon stomach (GAVE — gastric antrum vascular ectasia) kan gi akutt blødningsanemi og skyldes teleangiektasier i gastrointestinalkanalen. Gastroskopi gjøres for diagnostisering og i behandlingen brukes argon laser for koagulering (Ghrenassia E, 2014).
– “Stagnant loop syndrome” og pneumatosis cystoides intestinalis med luftbobler i tarmveggen er sjeldne manifestasjoner (Koysombat K, 2018).
-Anorektal dysfunksjon forekommer hos 50-70 % og ytrer seg ved obstipasjon, prolaps og anal sfinkter-dysfunksjon med fekal inkontinens (Garros A, 2017).
-Bakteriell overvekst diagnostiseres ved Glukose-pusteprøve. Pasienten drikker glukose, og man måler sa hydrogen (H) i utåndingsluften (kun bakterier kan produsere H).
Muskel/Skjelett
Artralgier er vanlig, men i praksis vanskelig å skille fra symptomer fra hud, underhud og sener.
Artritt kan forekomme og er erosiv i enkelte tilfeller.
Myopati med lavgradig myositt ses hos 14 % (kommer både sent og tidlig i forløpet) og særlig hos menn med diffus type. Obs! for Scleroderma-polymyositt overlapp syndrom (Skleromyositt). Karforandringer i neglesenger. Svinn av fingerpulpa (“Tuft resorption”) (Lefebreve F, 2021).
Tendon friction rub er seneaffeksjon med krepitasjoner ved bevegelse i seneskjeden. Manifestasjonen ses hovedsakelig ved diffus form og da tidlig in forløpet. Den kan indikere et komplisert sykdomsforløp (Barbachi A, 2023).
Andre manifestasjoner
Sekundært Sjøgrens syndrom, primær biliær kolangitt (PBC), autoimmun hemolytisk anemi og trigeminus nevralgi. “White matter hyperintense foci” kan påvises ved MR-cerebri.
Undersøkelser
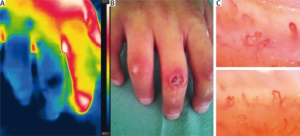
En tidlig mistanke om begynnende systemisk sklerose kan en få dersom det foreligger relativt nyoppståtte Raynauds fenomen, en påviser spesifikke antistoff som CENP eller anti-Scl70 i signifikante titre og kapillaroskopi er klart patologisk (se nedenfor). Grundige undersøkelser og noe oppfølging er likevel nødvendig for å sikre diagnosen og ikke minst vurdere prognostiske faktorer, samt sette opp et individuelt tilpasset behandlings- og oppfølgingsprogram (Volkmann ER, 2023).
Anamnesen kartlegger aktuelle symptomer og tegn på manifestasjoner. Dette er ganske omfattende (se nedenfor). Men en kan ta utgangspunkt i klassifikasjonskriteriene og etterspør Raynauds fenomen med ev. tid for debut, hovne fingre, sår på fingerpulpa, stramhet i huden på hender, ansikt og ellers på kroppen. Svelgevansker, magesyre-oppstøt/refluks, fordøyelsesbesvær, vekttap, tegn til dyspne eller tegn til perifere ødemer. Tørrhetsplager fra øye eller munn (sekundært Sjøgrens syndrom).
Klinisk kan en gjøre en generell status som kan omfatter måling av blodtrykk, puls og vekt. Auskultasjon av hjerte og lunger, palpasjon av abdomen. Huden på hender inspiserer og palperes for hovenhet/puffy, sklerodaktyli, sår eller substansdefekter på fingerpulpa, kalsinose og teleangiektasier. Kontrakturer beskrives. Ekstremiteter kan undersøkes for hud-manifestasjoner og bevegelighet.
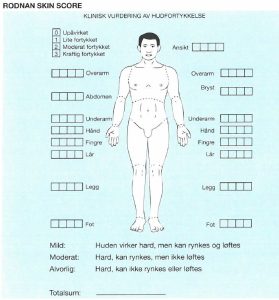
-Modifisert Rodnan Skin Score/hudskår (mRSS) har vist seg å korrelere med alvorlighetsgraden av hudmanifestasjoner. Metoden er blitt en gullstandard i evalering av hudtykkelse og utbredelsen ved systemisk sklerose. metoden er lett å lære, men en bør følge en prosedyre- pasienten skal være avslappet. Mellom undersøkerens tommel og pekefinger løftes huden forsiktig som beskrevet av Khanna (Khanna D, 2017).
Kapillaroskopi / kapillærmikroskopi / neglefold-video-kapillaroskopi. Et viktig hjelpemiddel i diagnostikken kan være kapillær-mikroskopi av neglesengene. Undersøkelsen kan integreres i en generell klinisk konsultasjon. Typisk vil disse vise dilaterte kapillærer, nedsatt tetthet av kapillærer, slyngede kapillære loops, avaskulære områder og «Bushy» fenomener (neo-angiogenese). Under kapillaroskopi kan det oppstå forbigående vasospasme i enkelte kapillærer som kan forveksles med manglende kapillærer (“ghost capillaries”). Tidlige forandringer: Mega-kapillærer med lekkasje av erytrocytter. Aktivt stadium: Spontan-hemoragier. Sent stadium: Kapillærtap, busk-kapillærer, få makro-hemoragier. Vennligst les mer om kapillaroskopi i eget kapittel
6-minutter gangtest. Det kan være nyttig med bestemmelse av avstanden som tilbakelegges i løpet av 6 minutters gangtid som både er en prognostisk indikator og et egnet middel til å evaluere effekten av behandlingen. Testen kan gjøres på en poliklinikk, dagenhet eller sengepost med hjelp av trenet personell. Den forutsetter imidlertid at pasienten har normal eller stabil gangfunksjon. Utenom lungene er også hjertefunksjonen avgjørende for resultatet (Enright PL, 2003).
Blodprøver. Rutineprøver kan omfatte CRP, SR, Hb, leukocytter med differensialtelling, trombocytter, elektrolytter, urat/urinsyre, lever-, nyre- og thyreoidea-funksjonsprøver. Glukose, kreatin kinase (CK), immunologiske prøver (se nedenfor) og urin stiks. Ved mulighet for pulmonal hypertensjon suppleres ofte med NT-pro-BNP.
–Ofte foreligger normale inflammasjons-parametere. Ved forhøyet SR og CRP kan det også foreligge det myositt med utslag i kreatin-kinase (CK). Vedvarende forhøyet CRP korrelerer også med IL-6 som og er ugunstige prognostiske parametere for lungefibrose, pulmonal hypertensjon og mortalitet (Mitev A, 2019). Immunologiske prøver: Se nedenfor.
Immunologiske undersøkelser. Ikke alle har utslag i antistoff.
- Antinukleære antistoff (ANA) positiv hos opp til 95%.
- Subgruppene Anti-Scl 70 (Anti topoisomerase I) hos 20-70 % (oftest initialt) av de med diffus type (humant DNA topoisomerase I er et enzym involvert i oppløsningen av vridningsstresset under DNA replikasjons-transkripsjon og kondensering av kromatin).
- Anti-centromer antistoff/CENP ses hos 40-75 % ved begrenset type (CREST).
- Anti-RNA polymerase III forekommer hos noen, hyppigst hos de som vil utvikle renal krise (SRC).
- Mer sjeldne auto-antistoffer er anti- Th/To (sensitivitet < 10 %), anti-U3RNP, U1RNP og anti-Ku.
- Opp mot 20% har primær biliær kolangitt (PBC) assosierte auto-antistoffer.
Lungefunksjonstester egner seg for å kartlegge om lungene er påvirket og for oppfølging for eksempel 1-2 ganger årlig i tilfelle ev. progresjon. Lungefunksjonstester: Intermediær: FVC > 70 = mild sykdom. Alvorlig: FVC < 70 = utbredt sykdom. Lungefunksjonstester er beskrevet i et eget kapittel.
Bildediagnostikk er viktige metoder både diagnostisk og i oppfølgingen av systemisk sklerose (Rutka K, 2021)
-CT (ev High Resolution /høyoppløsnings CT, HRCT) er den metoden som mest detaljert beskriver lungevevet og luftveiene (se også avsnittet Organmanifestasjoner nedenfor i dette kapitlet). Metoden gir også mindre stråledose enn vanlig CT. Et infiltrat (“consolidation”) defineres her som utvisking av karstrukturer og luftveisvegger, hvilket skyldes at alveole-luften erstattes av væske og celler. Ved mattglass-fortetning (“ground glass opacities”) foreligger det ingen utvisking. Mattglass-forandringer skyldes fortetninger intraalveolært og interstitielt. Arkitekturen er bevart. På et HRCT bilde skal man kunne skimte karstrukturene gjennom slike matte fortetninger. Dessverre er bare 20 % av slike forandringer reversible (resten er mikrofibrose). Bikake-forandringer (honeycombing) representerer endestadiet av en rekke lungesykdommer. Forandringene skyldes at intra-alveolære septa er fortykket, ødelagt eller anatomisk fortrengt. HRCT kan derfor anvendes for å beskrive de interstitielle forandringene nærmere. Imidlertid er ikke mattglass-forandringer alltid ensbetydende med pågående inflammasjon. Stadium-inndeling av CT forandringer (a.m. Athol Wells, Brompton Hospital): Utbredelse i lungevev omfatter < 20% mild sykdom. Utbredelse i lungevev > 20 % utbredt sykdom. CT kan også gi mistanke om pulmonal hypertensjon ved at diameter av pulmonalarterien er tydelig utvidet og videre enn aorta.
-Røntgen av øsofagus kan gjøres dynamisk med kontrastmiddel som pasienten svelger. En forventer redusert funksjon i distale 2/3 del av øsofagus. I blant påvises stenoser som kan behandles eller funksjonssvikt av musculus cricophayngeus i form av krikofaryngeal dysfunksjon som i noen tilfeller opereres (ØNH).
-Røntgen av hender gjøres hvis en vil vurdere om kalsinose foreligger. Ved kliniske mistanke og behov, undersøkes også andre kroppsdeler.
Bronko-alveolær lavage (BAL). En bronko-alveolær lavage ved Ssc hvor neutrofile overstiger 2.7 x 100 000/ml eller eosinofile over 2.3 % taler for inflammasjon. Den diagnostiske verdi av BAL er imidlertid omstridt. Noen har funnet at eosinofil alveolitt har en dårligere prognose enn neutrofil alveolitt. Vennligst les mer om BAL i kapittel om bronkoskopi.
EKG. Undersøkelsen kan avdekke tegn til kardial svikt eller pulmonal hypertensjon, arytmi eller iskemi.
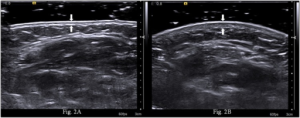
Ekkokardiografi / ultralyd Doppler gjøres ofte årlig over en lang periode. Hovedindikasjonen er å utelukke tegn til pulmonal hypertensjon. Tilstanden er overrepresentert både ved systemisk begrenset og diffus form for systemisk sklerose og behandlingsmulighetene klart bedre når en behandler tidlig.
Ultralyd av huden kan benyttes for å estimere hudaffeksjonen. Metoden har vist seg å korrelere med hudtykkelse, stivhet og Rodnan skin score (mRSS) (Liu H, 2017).
Hjertekateter-undersøkelse gjøres der ekkokardiografi tyder på pulmonal hypertensjon (PAH). PAH foreligger når høyresidig hjertekateter-måling viser mPAP ≥ 20mmHg i ro og motstanden i pulmonalarterien (PAR) er ≥ 3 Woods. Forutsetning er eksklusjoner: PCWP ≤15 mmHg (utelukker venstresidig hjertesvikt), kronisk lungesykdom med hypoksemi skal ikke foreligge, venøse tromber (tromboembolier) eller (sjelden) obstruktiv lungearterie-sykdom (tumorer, stenoser, parasitter) skal ikke foreligge, kronisk nyresvikt, sarkoidose, noen blodsykdommer og metabolske sykdommer som påvirker lungene skal ikke foreligge. Undersøkelsen og tolkning av resultater er beskrevet i kapitlet om pulmonal hypertensjon.
Andre. Enkelte (ikke rutine) bruker også økt fibrillin-1 som et aktivitetstegn på alveolitt ved SSc. Også 18FDG PET/CT scan kan brukes for å skille mellom inflammasjon og fibrose. Denne benytter seg av en isotop som ikke tas opp av fibroblaster. Positivt opptak tyder på inflammasjon i lungevev (tilstedeværelse av celler som metaboliserer glykose). Undersøkelsen er imidlertid ikke rutine. Transbronkial biopsi via bronkoskopi bør vurderes i tvilstilfeller. Termografi kan vise nedsatt temperatur ved Raynauds fenomen, men metoden er lite brukt i praksis.
Ulike typer systemisk skleroser

Begrenset Kutan Systemisk Sklerose. Hudaffeksjon distalt for albuer og knær, likevel forekommer ofte hudmanifestasjoner i ansikt og nakke, hyppig ILD (Interstitiell lungesykdom) og PAH (pulmonal hypertensjon). Nyreaffeksjon sjelden. Gjerne mangeårig Raynauds fenomen før utvikling av hudlesjoner). Vanligst antistoff er CENP (anti-centromer antistoff). CREST (Calcinosis/kalsinose, Raynauds, Esophagus/øsofagus-dysmotilitet, Sklerodaktyli og Teleangiektasier) er en form for begrenset SSc.
Diffus kutan systemisk sklerose. Hudaffeksjon på ekstremiteter, truncus og ansikt, samt hyppige organmanifestasjoner. Vanligste antistoff er Scl-70 (topoisomerase). Risiko for hypertensiv renal krise og GAVE (gastric antrum vascular ectasia), særlig de første to årene fra sykdomsdebut.
Juvenil systemisk sklerose er omtalt i et eget kapittel.
Pre-skleroderma og tidlig systemisk sklerose. “Udifferensiert systemisk bindevevssykdom med risiko for systemisk sklerose (UCTD-risk-SSc) / veldig tidlig systemisk sklerose” defineres ved Raynauds fenomen og enten autoantistoff forenelig med systemisk sklerose eller kapillaroskopi med sklerodermi-mønster uten at kriterier for systemisk sklerose er oppfylt. Aktuelle antistoff omfatter anti-ScL 70, anti-CENP, anti-RNA-polymerase III, anti-fibrillarin, anti-Th/To eller anti PM-Scl 70/100. Fordi 2013-ACR/EULAR kriterier ikke skal oppfylles, skal tilstanden ikke ha puffy/hovne fingre, sklerodaktyli, teleangiektasier, interstitiell lungesykdom eller pulmonal hypertensjon. Risikoen for å utvikle typisk systemisk sklerose i denne gruppen er 54% (Valentini G, 2020). Ved kombinasjonen av “puffy hands” og anti-Scl 70 eller anti-CENP antistoff er risikoen for utvikling av SSc særlig høy.
«Very early SSc»
Tidlige symptomer på systemisk sklerose som inngår i kriterier for tidlig Ssc Avouac J, Fransen J, ARD 2010:
- Raynauds fenomen
- «Puffy» / hovne fingre
- Typiske antistoff (Scl 70, CENP)
- Kapillaroskopi viser mikrovaskulær patologi
Vær oppmerksom på at ikke alle med «very early SSc» utvikler typisk SSc. En studie av 66 pasienter viste utvikling til SSc hos 32% etter 31 måneders oppfølging. Kapillaroskopisk megakapillærer var en risikofaktor (Vasile M, 2018;. Cutolo M, 2010).
Systemisk sklerose sine skleroderma. Typisk systemisk sklerose, men uten manifestasjoner i huden. Typisk hudaffeksjon tilkommer hos 60% i løpet av 0,5-7 år. Pasientene har ofte kliniske trekk som ved CREST syndrom. Disse pasientene kan utvikle pulmonal arteriell hypertensjon og asymptomatisk perikardsykdom, lunge, nyre, mage-tarm og ledd/muskel-manifestasjoner (LeRoy EC, 2001).
| Forskjeller mellom begrenset kutan systemisk sklerose og diffus kutan systemisk sklerose (Tilpasset etter Hinchliff M, 2008) | ||
| Kjennetegn | Begrenset form | Diffus form |
| Fibrose i huden | Fra distalt og opp til albuer og knær, kan affisere ansikt (rundt munnen) | Områder også proksimalt for albuer og knær. Kan affisere truncus, hals og ansikt |
| Typiske lunge-manifestasjoner | Pulmonal arteriell hypertensjon (PAH) | Interstitiell lungesykdom (ILD) |
| Karakteristiske indre organ-manifestasjoner | Alvorlig gastro-øsofageal refluks og Raynauds fenomen | Skleroderma renal krise |
| Kliniske funn | Teleangiektasier, kutan kalsinose, sklerodaktyli, digitale iskemiske komplikasjoner (sår, nekroser) | “Tendon friction rub” (sene-krepitasjoner), pigmentforandringer |
Skleromyositt. Skleroderma-myositt overlapp. Dette er en egen sykdomsenhet. Hovedsymptomer er Raynauds, skleroderma-forandringer i hud i ansikt og på hender, artralgi eller arteritt, myositt og interstitiell lungesykdom (nær 100%). Andre organer angripes sjelden og forløpet er med godartet enn skleroderma diffus form. Påvisning av anti-PM Scl 75 eller 100 er typisk. Myokarditt, ofte asymptomatisk er ikke helt sjelden (18%) (Lilleeker JB, 2017). Behandlingen kan ved behandlingskrevende myositt bestå i lave doser prednisolon, høyere doser unngås på grunn av risiko for renal krise. Litteratur: Bhansing KJ, 2014; Pope JE, 2002.
Barnetts klassifikasjon (Noen foretrekker fremdeles Barnetts klassifikasjon): Type I: affeksjon av utelukkende fingre. Type II: affeksjonen rammer også underarmer. Type III: diffus affeksjon.
Diagnosen systemisk sklerose (systemisk begrenset eller diffus form) stilles på typisk anamnese, sykdomsbilde med Raynauds fenomen, sklerodaktyli og auto-antistoffer. Typiske forandringer ved kapillaroskopi forventes også. Hudbiopsi er sjelden nødvendig. ANA forventes å slå ut i blodprøver, og CENP eller a-Scl70 er typiske subklasser.
Klassifikasjonskriterier
ACR/EULAR-kriteriene av 2013 for klassifikasjon krever enten proksimal skleroderma eller to av følgende: sklerodaktyli, digital iskemi eller pulmonal fibrose (se tabellen nedenfor)
| Klassifikasjonskriterier. 2013 (van den Hoogen F). Sum-score på minst 9 for diagnose: | Score (vekting): |
| Sklerodaktyli på fingre og proksimalt for MCP bilateralt | 9 |
| Puffy fingre | 2 eller |
| Sklerodaktyli av minst en hel finger distalt for MCP | 4 (velg høyeste skår) |
| Ulcera på fingerpulpa | 2 eller |
| Pitting scars/arr/skorper på fingertupp | 3 (velg høyeste skår) |
| Telangiektasi | 2 |
| Kapillaroskopi patologisk | 2 |
| Pulm hypertensjon eller/og interstitiell lungesykdom | 2 |
| Raynauds fenomen | 3 |
| Antistoff relatert til systemisk sklerose (CENP, ScL70, RNA Polymerase III) | 3 |
Differensialdiagnoser
- Acrodermatitis chronica atrophicans (kronisk borreliose)
- Amyloidose (ikke-inflammatorisk hovne hender og glatt hud)
- Buschkes syndrom (sklerødema adultorum Buschke)
- Diabetisk hånd. Ved mangeårig diabetes. Ikke Raynauds
- Eosinofil fasciitt. Hendene spares. Eosinofili i blod og/eller vev.
- Fasciitt-pannikulitt syndrom. Tidligere oppfattet som subgruppe av eosinofil fasciitt, men uten eosinofili. Ofte kreft-relatert.
- GVHD Kronisk graft versus host disease
- Huriez syndrom (palmoplantar keratodermi med sklerodaktyli)
- Kjemisk indusert sykdom
-
- Acro-osteolyse kjennetegnes ved Raynauds fenomen og smertefull hevelse av de distale falangene, samt subkutan kalsifikasjon, evt. sakroiliitt og assosiasjon til vinylklorid eksposisjon.
- Eosinofili myalgi syndrom: Inntak av 1-tryptofan. Epidemi i Mexico i 1989. Ingen Raynauds.
- Erasmus syndrom. SSc etter eksposisjon for silika (brukes i betong-produksjon).
- Kjemisk / medikament / rusmiddel -induksjon ved vinylklorid, bleomycin, pentacozin, Vitamin B12, Vitamin K, kokain, penicillamin, methyrsergid
- Nefrogen fibroserende syndrom: Gadolinium i kontrastvæske. Hos pasienter med nedsatt nyrefunksjon.
- Toxic oil syndrome (inntak av rapsolje kontaminert av fenylamino propanediol). Epidemi i Spania i 1981. Ingen Raynauds, men ellers lik SSc.
-
- Lichen sclerosus et atrophicus kan affisere alle deler av kroppen, men oftest genitalier (vulva, penis). Hvite hyperkeratotiske flekker som kan klø, blø og medføre smerter.
- Lipodermatosklerose (pannikulitt subkutant i begge legger. Harde legger proksimalt for ankler. Eldre personer. Ukjent årsak)
- Lokalisert sklerodermi (Morfea: ulike typer)
- Nefrogen systemisk fibrose (NSF). Etter gadolinium i kontrastvæske. Hos pasienter med nedsatt nyrefunksjon.
- Mediastinal fibrose / fibroserende mediastinitt
- Melorheostosis. Begynner i barnealder. Fortykket, lokalisert benstruktur. Kan medføre smerte, fysisk deformitet, hud og sirkulasjons-problemer, kontrakturer og redusert fysisk funksjon
- Morfea (Lokalisert skleroderma)
- Lineær sklerodermi (“coup de sabre”).
- Keloid morfea (irregulære smertefulle noduli).
- Generalisert morfea
- Andre typer
- Nodulær sklerodermi. En meget sjelden tilstand som gir seg til kjenne ved multiple keloid-liknende lesjoner. Pasientene har ofte artralgier, sklerodaktyli, Raynauds fenomen, digital pitting, kalsinose og lungesykdom. Mer sjelden er pulmonal arteriell hypertensjon, nyresykdom. SR ofte normal.
- Peyronies sykdom (fibromatose i penis)
- POEMS syndrom (polynevropati, organmegali, endokrinopati, monoklonal gammopati, hyperpigmentering og hudfortykkelse)
- Poikilodermi (inflammatorisk eksem fra barnealder, non-cyklisk neutropeni, luftveissymptomer, negledystrofi, hyperkeratose, kalsinose, kortveksthet) (Wang L, 2017).
- Porfyri (curana tarda). Arr-forandringer etter multiple hudskader på lys/sol-eksponerte områder
- Progeria (Hutchinson-Gilford syndrom, Werners syndrom) begynner i to års alder. Tidlig alderdom, fugle-ansikt, tynne ben
- “Puffy hands” av andre årsaker: Psoriasisartritt, MCTD, amyloidose
- Retroperitoneal fibrose og multifokal idiopatisk fibrosklerose (IgG4 relatert sykdom).
- Raynauds fenomen av andre årsaker: Andre systemiske bindevevssykdommer. Primær Raynauds
- Skleromyksødem (papulær mucinose)
- 2-4mm store papler, til dels med flat overflate. Inneholder mucin, ikke puss. Grupper og lineær utbredelse over dorsalside av hender, ansikt, albuer og ekstensor-sider er typisk. Utbredelse over større deler av kroppen kan gi et systemisk sklerose/sklerodermi-lignende bilde med reduserte bevegelsesutslag (Hummers LK 2014).
- Sklerødem Buschke
- “Stiff skin syndrom”: Starter ofte i barne- og ungdomsårene. Ingen organaffeksjon. Histologi viser ingen inflammasjon. (Guiducci S Rheumatology 2009)
- Venøs insuffisiens, kronisk med sekundær dermato-sklerose
Svangerskap ved SSc
Svangerskap kan ofte gjennomføres, men bør generelt frarådes ved alvorlig organmanifestasjon som alvorlige manifestasjoner i lunger, nyrer eller hjerte. Pasienter med aktiv progredierende diffus form har størst risiko for svangerskapsrelaterte komplikasjoner, til tross for tett, multidisiplinær oppfølging (Braun J, 2022). Data er usikre fordi aktuelle studier er små med risiko for seleksjons-feil, men generelt vil i løpet av svangerskapet omtrent 25 % bli bedre, 15 % verre, 60 % uendret og 35 % forverres post partum. -Økt risiko foreligger for preeklampsi, intrauterin veksthemming, prematuritet og spontane aborter. En av årsakene kan være vaskulopati i placenta (Ibba-Manneschi L, 2010). -Renal krise kan ses i ca. 2% av svangerskap ved systemisk sklerose. Det er essensielt å skille denne fra preeklampsi og eklampsi. Renal krise kan opptre når som helst, men særlig ved diffus form av systemisk sklerose og de første to årene fra diagnose. Behandling med ACE-hemmere kan gi fosterskader – særlig hvis de brukes i tredje trimester. På vital indikasjon kan ACE hemmer likevel være nødvendig ved renal krise (Samaritano LR, 2020). Alternativer er andre antihypertensiva. Uansett bør fosteret kontrolleres for utvikling av oligohydramnion. Generelt om svangerskap ved revmatisk sykdom i eget kapittel. Vennligst se også info fra NKSR.
Sjekkliste når svangerskap ved systemisk sklerose blir planlagt:
- Forbudte medikamenter seponeres.
- Tegn til organskader må bli vurdert før graviditet.
- Pulmonal hypertensjon er en kontraindikasjon.
- Lungefunksjon (spirometri, CT).
- Hjertefunksjon (EKG), ekkokardiografi.
- Nyrefunksjon (blod og urinprøver).
- Sjekk vitamin D og jern-status (25-OH vit-D, transferrin-reseptor). Mulig redusert opptak fra tarmen ved systemisk sklerose kan føre til for lavt vitamin D i blodet.
- Antistoff mot ScL-70 og RNA-polymerase III øker komplikasjonsfaren ved graviditet.
- Antifosfolipid-antistoff bør sjekkes selv om økt forekomst ikke forventes ved systemisk sklerose.
- Gynekologisk vurdering overveies.
- Generell informasjon om sykdommen og om graviditet.
Sjekkliste under svangerskap ved systemisk sklerose:
- Følges opp som et «risikosvangerskap» i samarbeid med fødepoliklinikk (fosterets vekst og blodsirkulasjon, sjekker tegn til for tidlig fødsel).
- Medikamenter som tillates (om nødvendig): Protonpumpehemmere (mot magesyreoppstøt), kalsium-antagonister (Adalat) (mot høyt blodtrykk eller Raynauds fenomen/likfingre), antihistaminer (mot allergi og sterk klør), lav dose acetylsalisylsyre (Albyl-E 75-160mg, fra svangerskapsuke 12) (Forebyggende mot pre-eklampsi). Prednisolon eller andre kortikosteroider unngås eller brukes i lavest mulig dose.
Sjekkliste ved fødsel og systemisk sklerose:
- Informer fødestuen dersom huden er preget av sykdommen; Kan gi problemer for anestesi og for å finne venøs tilgang.
- Regional anestesi (spinal/epidural) foretrekkes fremfor narkose (intubasjon).
- Etter fødsel: Følg blodtrykket.
- Episiotomi eller sår etter sectio gror vanligvis ukomplisert.
Behandling
Fordi systemisk sklerose angriper mange organer kan ikke en behandlings-algoritme anvendes på alle pasienter. Det må alltid gjøres individuelle, unike tilpasninger. I tillegg må en regne med å gjøre justeringer under veis ut i fra forløpet av den underliggende sykdommen, intoleranse for behandling eller ny komorbiditet. For eksempel kan sykdommen medføre betydelig underernæring med økt risiko for opportunistiske infeksjoner, særlig blant dem som får immundempende medikasjon. Inntak av forskrevne medikamenter kan hindres av øsofagusdysmotilitet og sykdommen disponerer for depresjon (Thoms BD, 2007) som også kan påvirke compliance/etterlevelse (Volkmann ER, 2023). En har prøvd ut en rekke ulike potensielt sykdomsdempende medikamenter for å stanse og reversere systemisk sklerose, men med begrenset effekt. Samarbeid med et adekvat spesialisert senter med godt kjennskap til sykdommen kan være viktig (Christopher P Denton, Dinesh Khanna, Lancet 2017).
Behandlingen består av:
- Generelle tiltak for å forebygge progresjon og komplikasjoner
- Behandling av truende organ-komplikasjoner
- Immun-supprimerende medikamenter (i noen tilfeller)
Digitale sår: Varmehjelpemidler, kalsiumblokker, phosphodiesterase 5 hemmer (sildenafil), topisk nitrat (nitroglyserin salve), Ilomedin iv eller Botox injeksjoner, endotelin reseptor-antagonist (bosentan med flere). Generelle tiltak: Unngå infeksjon og forfrysning, bruk hansker (ev med varmetråder) og lue. Fysioterapi, øvelser med øvelser for munn og hender, samt aerobisk trening er vist å kunne styrke muskler og bedre pasientenes daglig funksjon. Fordi sykdommen er sjelden og individuelt forskjellig, vil fysioterapeuter oftest trenge spesielt gode henvisninger fra lege som kjenner sykdommen og den aktuelle pasientens behov (Liem SIE, 2022).
Gastrointestinale manifestasjoner: Refluksøsofagitt behandles med syrepumpehemmeren esomeprazol (Nexium 20-40 mg/d) eller pantoprazol (Somac). Medikamentet bindes i parietalcellen til H+, K+ ATPase molekylet som kalles syrepumpen). Viktig er også opphøyd hodeleie, inntak av store måltid midt på dagen, unngå måltider sent på kvelden. Ved strikturer bør kirurg konsulteres. GAVE, bakteriell overvekst, anal inkontinens, malabsorpsjon og pseudoobstruksjon behandles i samråd med eller etter anbefaling av gastroenterolog. Ved bakteriell overvekst i tarmen brukes vekslende antibiotika-regimer. Ved obstipasjon er tilpasset kosthold spesielt aktuelt. Motilitetsfremmende medikamenter brukes i enkelte tilfeller. Fekal inkontinens kan utredes og følges nærmere opp via gastroenterolog.
Hjerte-manifestasjoner utredes og behandles i samarbeid med kardiolog. Fosfodiesterase 5 (PDE5)-hemme (oftest sildenafil), endotelinreseptor-antagonister (ambrisentan, bosentan) eller prostasyklin reseptor-angonist (seleksipag) er aktuelle, til dels i kombinasjoner. Ved relatert kardial svikt brukes bumetanid (Burinex) ofte. Pulmonal hypertensjon er omtalt i eget kapittel.
Huden kan ha nytte av fuktighetskremer og øvelser for å opprettholde elastisitet (fleksjon- og ekstensjon av fingre, smile- og grimase-øvelser for ansikt). Immunsuppressive medikamenter som mykofenolat kan ha en viss effekt og vurderes individuelt, oftest ved diffus, progredierende form. Ster kløe kan lindre med antihistamin. Teleangiektasier kan enkeltvis behandles med laser på kosmetisk indikasjon av hudlege. Kalsinose kan fjernes kirurgisk, men residiverer hyppig. Mot karpemunn er Restylan brukt kosmetisk i enkelte tilfeller.
Lungesykdom. Ved tegn til lunge-inflammasjon vurderes for DMARDs. De mest aktuelle er mykofenolat (MMF) og cyklofosfamid. Også anti-fibrotisk behandling med nintedanib (Ofev) kan vurderes. Ved manglende effekt eller intoleranse skiftes mellom de ovenfor nevnte eller rituksimab eller lungetransplantasjon. Alvorlig, raskt progredierende lungesykdom kan være medvirkende indikasjon for HMAS (høydose kjemoterapi med autolog stamcellestøtte) ved diffus form av systemisk sklerose. Behandling med tocilizumab eller azathioprin ev andre DMARDs kombinert med kortikosteroider er omdiskutert (Hoffmann-Vold A, 2022). Dersom blodgass er lav (SpO2 < 95%), kan oksygentilskudd ved fysisk aktivitet og under flyreise være aktuelt, noe som vurderes av lungelege. Ved redusert lungefunksjon bør en alltid overveie om lunge-rehabilitering er indisert. Studier og klinisk erfaring har tydelig vist nytten av slike tiltak (Castro AAM, 2013).
Myositt ved skleromyositt (se ovenfor) kan en lav dose prednisolon (<10mg/dag for å unngå hypertensiv nyrekrise og redusere steroid-bivirkninger). Metotreksat kan også være aktuelt.
Nyrekrise (SRC). ACE-inhibitorer blokkerer overgangen angiotensin I til angiotensin II. Angiotensin I og renin fortsetter å akkumuleres, men er ikke biologisk aktive. ACE-hemmere proteolyserer også bradykinin som er en viktig vasodilatator. Sirkulatorisk stabile pasienter kan bruke ramipril initialt 5 mg, med økende dose til 10 mg/d. Et alternativ er captopril, initialt 6,25 mg – 12,5 mg, ACE- hemmere kontinueres også under dialyse, selv i små doser. Gjelder også normotensive SRC. Målet er BT 120/70-80. Fortsett også etter normalisering av nyrefunksjon hvis BT må behandles. Vurder ikke transplantasjon for etter 18 måneder.
Raynauds fenomen. Kalsiumblokker (nifedipin -Adalat® Oros, amlodipin -Norvasc® ) (OBS! hypotoni). Sildenafil er et alternativ.
NSAIDs er en tilbakeholden med på grunn av bivirkningsrisiko ved svelgevansker, noen har GAVE og dysmotilitet i tarm er vanlig.
Immundempende behandling: -Kortikosteroider kan bidra til å utløse renal krise, særlig ved diffus form. Doser over 15 mg/d øker risikoen betydelig. -csDMARDs, oftest mykofenolat, brukes i noen tilfeller for å redusere progresjon av hud- og lungeforandringer. Metotreksat kan være aktuell mot artritt. -Biologiske medikamenter som forsøkes (utprøvende) mot hud og lunge-manifestasjoner er B-celle hemmere (rituksimab), anti-interleukin-6 hemmer (tocilizumab). -Janus kinase hemmere (JAK-hemmer) og transforming growth factor β hemming som utprøvende behandling har vist en viss effekt på hud og lungemanifestasjoner. -HMAS (Høydose kjemoterapi med autolog stamcellestøtte). Vennligst se eget kapittel om HMAS. –CAR-T cellebehandling. Kimerisk antigenreseptor-T-celleterapi (CAR-T cellebehandling) er et nytt, svært potent behandlingsprinsipp under utprøving i revmatologi. Metoden benytter genteknologi til å modifisere T-celler slik at de gjenkjenner spesifikke antigen på B-cellers overflate. Det er rapportert lovende effekt av utprøvende CAR-T cellebehandling ved SLE og myositt, men også ved systemisk sklerose Bergmann C, 2023.
Anbefaling: Norsk Revmatologisk forening / legeforeningen har utarbeidet en detaljert veileder for behandling ved systemisk sklerose.
Kontroll og oppfølging av SSc
| Et forenklet kontroll-opplegg (minimum) er foreslått for pasienter med tidlig sykdomsform (se ovenfor). Ved patologiske organ-funn ved diagnose skal disse følges opp i tillegg (Gonzalez-Garcia A, 2022). |
|
| Ved diagnose | Årlig kontroll |
| Røntgen øsofagus eller manometri | |
| Lungefunksjonstester inklusiv DLCO
HRCT NT-ProBNP Doppler sonografi/ekko cor |
Lungefunksjonstester inklusiv DLCO |
| EKG | Doppler sonografi/ekko cor |
Regelmessig kontroll, for eksempel årlig, bør gjøres av følgende:
- Hud. Forløpet følges med Rodnan hudscore
- Nyrer. Nyrefunksjonen måles ved kreatinin eller GFR. Blodtrykk.
- Hjertet. Ekkokardiografi med tanke på pulmonal arteriell hypertensjon, årlig de første årene fra sykdomsdebut. PRO-BNP i blodet (øker ved pulmonal hypertensjon)
- Lunger: Ikke stol på pasientens angivelse av manglende funksjons-dyspné. Mange av disse pasientene er såpass funksjonshemmet at de ikke belaster og derved ikke merker en eventuell dyspné.
- HRCT av lunger regelmessig i en begrenset periode for a avdekke ILD og lungecancer. Det er imidlertid holdepunkter for at pasienter med normal HRCT og normale lungefunksjonstester de første sykdomsårene har meget liten risiko for å utvikle ILD senere. Oppfølging av slike pasienter bør derfor baseres på klinikk alene.
- Lungefunksjonstester kan delvis erstatte CT i forløpet.
- 6-minutter gangtest
- Malignitet-screening er ikke rutine ut over det som anbefales befolkningen generelt. Generelt disponerer heller ikke systemisk sklerose for kreft. Et unntak kan gjelde pasienter som nylig er diagnostisert og har anti-RNA polymerase 3 antistoffet (Lazzaroni M-G, 2017)
Prognose
Diffus form av systemisk sklerose er den mest alvorlige blant de systemiske bindevevssykdommene, og det er påvist økt dødelighet. Tidligere diagnose og bedre oppfølging de senere år forventes imidlertid å bedre utsiktene. Historiske data indikerer at grunnsykdommen er årsak til 55% av alle dødsfall, hvorav 35% skyldes ILD, 26% PAH og 26% hjerteaffeksjon, men det er store individuelle forskjeller. Jo mer proksimal hudaffeksjon (diffus form), desto dårligere utfall. Ved sklerodaktyli alene er 10 års overlevelse 71 %, ved hudaffeksjon proksimalt for MCP 58% og ved engasjement av truncus 21 %. Utvikling av hudfortykkelse før opptreden av Raynauds fenomen indikerer alvorlig sykdom. Mangeårig Raynauds før hudmanifestasjoner kan tyde på benignt forløp. Hjerteaffeksjon signaliserer dårlig prognose. Nyre- og lungeaffeksjon bidrar vesentlig til forverret prognose. Ved pulmonal arteriell hypertensjon og 6 minutters gangtest under 330 m er prognosen dårlig. Ved stigende Pro-BNP hos pasient med PAH er dødeligheten meget høy. 1, 2, 3 og 4 års overlevelse etter diagnosen PAH er 86, 59, 39 og 22%. 1, 3 og 5 års overlevelse ved SSc-ILD er 100, 90 og 77% (Tyndall AJ, 2009). Vedvarende forhøyet CRP korrelerer også med IL-6 som og er ugunstige prognostiske parametere for lungefibrose, pulmonal hypertensjon og mortalitet (Mitev A, 2019).
Tidligere studier har vist økt insidens av cancer i lunge, mamma, tunge og hematologisk malignitet (Szekanecz E, 2012), men det er uklart om årsaken er miljøfaktorer inklusiv tidligere behandling med cyklofosfamid eller om grunnsykdommen er disponerende (Sargin G, 2018).
Retningslinjer, anbefalinger og prosedyrer
EULAR: Parodis I, 2023 (non-farmakologisk behandling)
EULAR: Kowal-Bielecka O, 2016 (management)
EULAR/EUSTAR: Walker KM, 2011 (management)
Kanada: Pope J, 2012 (Management)
Norsk Revmatologisk forening / legeforeningen

{kind=link}