BINDEVEVSSYKDOMMER (REV 021-033)
58 MCTD Mixed connective tissue disease – Blandet bindevevssykdom (REV 021)
MCTD, Blandet bindevevssykdom
Ragnar Gunnarsson
Kjennetegn på MCTD
Raynauds fenomen, hovne (“puffy”) fingre er tidlige symptomer.
Overlappende symptomer med andre bindevevssykdommer som lavgradig myositt, sklerodaktyli (systemisk sklerose) og eksantem (SLE) er også typisk.
ANA er oftest positiv og sub-gruppen RNP er obligatorisk.
MCTD er en sjelden autoimmune bindevevssykdom med prevalens på rundt 4 per 100.000 og insidens fra 1,8 – 0,2 per 100.000 per år. MCTD rammer kvinner 3-6 ganger oftere. Gjennomsnittsalderen for å få MCTD diagnose er mellom 30-40 år.
De aller fleste pasientene som har fått MCTD diagnose utvikler ikke en annen systemisk bindevevssykdom. I en nylig publisert norsk oppfølgingsstudie var det kun 9,5% (14/147) som utviklet en annen systemisk bindevevssykdom.
Det finnes fire kriterier for MCTD. Alarcon-Segovia og Kasukawa kriteriene brukes mest i kliniske studier. Sharps modifiserte kriteriesett er for komplisert for klinisk bruk og Kahns kriteria har vært svært lite brukt.
MCTD er assosiert til risiko allelene; DRB1*04:01 og HLA B*08 og skiller seg fra systemisk lupus erythematosus, systemisk sklerose og inflammatoriske myositter. Høytitrete antistoffer rettet mot 70 kDa antigenet i U1-nukleære ribonukleoprotein-partikkelen (U1 snRNP) i perifert blod, sammen med en rekke spesifikke kliniske funn, er nødvendig for diagnostisering av MCTD.
De spesifikke kliniske symptomer og funn for MCTD er trefasiske Raynauds fenomen, diffust håndødem (“puffy hands”), myositt, leukopeni, spiserørs dysmotilitet, pleuritt, perikarditt, interstitiell lungesykdom, og pulmonal hypertensjon.
Definisjon
Mixed connective tissue disease» (MCTD) er en sjelden systemisk autoimmune bindevevssykdom. Dr. Gordon C Sharp lanserte konseptet MCTD, som har nå vedvart i nesten femti år (1).
Høytitrete antistoffer rettet mot U1-nukleære ribonukleoprotein-partikkelen (U1 snRNP) i perifert blod sammen med en rekke kliniske funn, er nødvendig for diagnostisering av MCTD. MCTD diagnosen bør vurderes hos en pasient med anti-RNP antistoffer og som har trefasiske Raynauds fenomen, diffust håndødem (“puffy hands”), og har eller har hatt noe av følgende; artritt, myositt, leukopeni, spiserørs dysmotilitet, pleuritt, perikarditt, interstitiell lungesykdom (ILD), eller pulmonal hypertensjon (PH). Ingen av disse kliniske funnene er eksklusive for MCTD (2). Sykdommen MCTD har kliniske trekk som kan ligne på andre autoimmun sykdommer som systemisk lupus erythematosus (SLE), systemisk sklerose (SSc) og inflammatoriske myositter (polymyositt/dermatomyositt (PM/DM)) (2).
Det er ingen internasjonal enighet om hvordan og i hvem MCTD skal diagnostiseres. Det finnes fire kriterier for diagnostisering av MCTD. Disse ble opprinnelig presentert som diagnostiske og klassifikasjons kriterier. De er dels forskjellige og fanger de ikke helt de samme pasientene. Bare to av disse fire kriteriene har blitt brukt jevnlig til forskningsformål: Alarcon-Segovia og Kasukawa-kriteriene (3, 4). Sharps modifiserte kriteriesett har ikke blitt brukt mye av flere årsaker, det mest åpenbare er deres kompleksitet og behovet for antistoff analyse som ikke lenger er i bruk (5). Kahn-kriteriene de ble opprinnelig presentert på fransk, og det faktum at de lignet et av de allerede publiserte kriteriesettene gjør at det er svært lite brukt (6).
I en landsomfattende norsk kohortstudie var prevalensen av MCTD lik på tvers av de tre kriteriene for MCTD, men Kahn kriteria var der ikke brukt pga. likhet med Alarcon-Segovia kriteria. Det store flertallet av pasientene i Norge (97%) oppfylte minst to av de tre MCTD-kriteriene og 75% oppfylte alle de tre anvendte kriteriene (7). Det var imidlertid forskjeller, spesielt i forhold til pasienter med ILD, der de ikke vil bli fanget opp av verken Alarcon-Segovia (eller Kahn) kriterier (Figur 1).
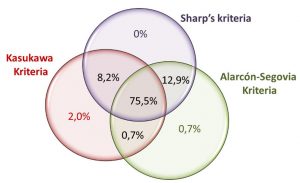
Epidemiologi
. CC BY-NC-SA 3.0")
Det er få epidemiologiske studier på MCTD. MCTD er sjeldnest av de kjente systemiske autoimmune bindevevssykdommer, som SLE, SSc, PM/DM og primært Sjögrens syndrom (pSS).
En nylig publisert studie fra Olmstad fylke (pop. 150.000) i delstaten Minnesota i USA, fant 50 pasienter på 30 år fra 1985 tom 2014 og ut fra det estimert annuell insidens på 1,9 per 100.000. (8). Prevalens av MCTD i en landsdekkende norsk tverrsnittsstudie, var på 3,8 per 100.000 i 2008. Retrospektiv estimerte insidens var betydelig lavere på 2,1 (per million per år) (7).
MCTD har det felles med de andre autoimmun bindevevssykdommene, at sykdommen rammer kvinner hyppigere. Av de som får MCTD er 70-85% kvinner. I Norge var MCTD 3,3 ganger hyppigere hos kvinner (70%) og i Olmstad fylke (USA) var 5,3 flere kvinner (84%) som fikk MCTD diagnosen (7, 8). Sykdommen rammer alle aldersgrupper, det er svært sjelden at barn i førskolealder får diagnosen og aller fleste som får MCTD i ung alder får dette i ungdomsårene. Gjennomsnittsalderen for pasientene ved den første sykdoms manifestasjonen i Norge var 31,5 år, mens diagnosen MCTD var ved gjennomsnittsalderen 35 år, mens 10% fikk diagnosen før de fylte 18 års alder (7). I Olmstad fylke, USA så man kun på de som fikk diagnosen etter 18 års alder og var gjennomsnittsalderen betydelig høyere på 48 år (8). Illustrasjon: Sen S, Sinhamahapatra P, Choudhury S, Gangopadhyay A, Bala S, Sircar G, Chatterjee G, Ghosh A – Indian journal of dermatology (2014). CC BY-NC-SA 3.0
Før trodde mange at de MCTD var et tidlig tegn på en annen systemisk bindevevssykdom og sykdommen ville deretter forandre seg til en av de andre tre andre sykdommene; SLE, SSc PM/DM. Nyere studier viser en annen virkelighet der aller fleste pasientene vedvarer som MCTD (9, 10).
Genetikk
HLA DRB1* 04:01 er hovedrisiko-allel for MCTD. MCTD er også assosiert til HLA-B* 08 (11-15). HLA-profilene til MCTD på gruppenivå skiller seg tydelig fra HLA-profilene til etnisk matchede friske kontroller, og til pasienter med SLE, SSc og PM/DM.
Patogenese
Det er ukjent hvordan genetikk, med risikoallelene HLA B*08 og DRB1*04:01 bidrar til dannelse av anti-RNP autoantistoff eller sykdomspatogenese. Det generelle scenariet for utvikling systemiske bindevevssykdommer, er at personer med predisponerende genetisk bakgrunn, utsettes for miljømessige risikofaktorer og deretter utvikler systemisk, kronisk immunaktivering. Denne hypotesen er også relevant for MCTD, men det foreligger foreløpig ingen konklusive data om risikofaktorer.
Høye antistoff titer rettet mot U1-smånukleære ribonukleoprotein-partikkelen (U1 snRNP), på et tidspunkt, er nødvendig for diagnosen MCTD. Biokjemiske analyser har vist at anti-RNP-antistoffene binder tre U1-spesifikke proteiner A, C og 70 kDa i det makromolekylære U1-snRNP-komplekset. Tilgjengelige data indikerer at 70 kDa-antigenet er hovedmålet for serum-anti-RNP-antistoffer i MCTD (16-18) (Figur 2). U1-snRNP komplekset inneholder mange andre antigener målrettet av CTD-assosierte auto-antistoffer, inkludert anti-Smith (Sm).
Rollen til anti-RNP-antistoffene i patogenesen av MCTD forblir ukjent, men nylige data har forsterket forestillingen om at auto-antistoffproduksjon kan bli genetisk bestemt og drevet av distinkte undergrupper av HLA-begrensede T-celler. Dessverre er dataene motstridende. Hoffman (19) og Smolen og kolleger (20), rapporterte sammenheng mellom anti-RNP antistoff og vevstypen HLA-DR4 ved SLE, mens Olsen og kolleger fant ingen slik sammenheng (21).
Data fra HLA-transgene mus med MCTD-lignende sykdom antydet at anti-RNP-antistoffene kan være sykdomsfremkallende (21), men det er ingen data om dette hos mennesker. Så vidt vi vet er det ingen studier om den patogene rollen til anti-RNP antistoff ved andre autoimmune systemiske bindevevssykdommer.
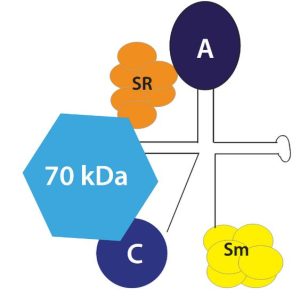
Symptomer
Raynauds og “puffy hands”
-Raynauds fenomen (RP) ble først beskrevet av Dr. Maurice Raynaud i 1862 og forekommer i en primær og sekundær form (22). RP er anfallsvis tilstand indusert av lav temperatur med vanligvis tre faser: avblekning, cyanose og reaktiv hyperemi. Minst to av tre symptomer bor være til stede for det kan kalles Raynaud fenomen. Symptomene kan bli sa uttalte at det minner om en kronisk cyanose. Sekundær RP er assosiert med; mekaniske, kjemiske eller strukturelle endringer i karveggene. Sekundær RP sees ved flere systemiske bindevevssykdommer, men er mest utbredt i SSc og MCTD (23). Overveldende flertall av pasienter med MCTD har RP. I det landsdekkende norske kohorten (7) ble RP rapportert hos 99% av pasientene; sammenlignbar med tidligere studier (24-26). Det har blitt rapportert at pasienter med sekundær RP på grunn av SSc, MCTD og DM har karakteristiske strukturelle forandringer i negleseng-kapillærene (27-31). Dessverre har ikke negleseng-kapillaroskopi-mønstrene for de forskjellige CTD-ene blitt systematisk sammenlignet. Et større MCTD-kohort viste at 38% hadde scleroderma kapillaroskopi-mønster (32).
-Puffy hands. Hovne hender/fingrer eller det som kalles “puffy hands” er blant de vanligste sykdoms manifestasjonene. Mekanismene bak den symmetriske hevelsen i hånden er ikke tydelige, men tenosynovitt og / eller endotelcelledysfunksjon er foreslått (33). MCTD-kohortstudier har rapportert “puffy hands” hos 60-94% (24, 25, 34).
Myositt er inkludert som et av hovedkriteriene i alle de fire kriteriesettene for MCTD. Tilgjengelige data indikerer at 35-79% av MCTD-pasienter vil utvikle tegn på myositt i løpet av sykdomsforløpet (7, 24-26, 34-36). Det ser ut til at MCTD-assosiert muskelinvolvering har samme distribusjon som ved inflammatorisk myositt (PM/DM) men ser ut til å forårsake mindre permanent skade (37). Data om muskelhistologi ved MCTD er begrenset, men histologisk er det funn forenlig med dermatomyositt (DM). I en stor europeisk myositt kohort (38) ble auto-antistoffer mot anti-U1snRNP antistoffer rapportert hos 6% (25/417). Det ble ikke rapportert om noen av de anti-RNP-positive pasientene fikk diagnosen MCTD. De anti-RNP positive pasientene i kohorten ble behandlet med gjennomsnittligere lavere steroiddose og så ut til å ha en bedre prognose angående muskelinvolvering en de øvrige pasientene.
Symptomer fra ledd er vanlig assosiert til MCTD (39), og artritt er inkludert i alle de fire MCTD kriteriene. Dessverre har man ingen gode studier på leddaffeksjon ved MCTD Det vanligste mønsteret for leddaffeksjon ser ut til å være polyartritt. I en studie fra 1977 ble destruktiv (erosiv) leddaffeksjon beskrevet i hele 53% av MCTD-tilfellene (40, 41).
ILD er en av de alvorligste sykdomskomplikasjonene ved MCTD (25, 26, 42-46). Tverrsnittsdata her fra Norge viste at 52% av pasientene som ble undersøkt (n = 126) hadde unormale funn på høyoppløselig CT av lungene, oftest retikulære mønster forenlig med lungefibrose (35%). Lungefibrosen ble kvantifisert som lavgradig hos 7%, moderat hos 9% og høygradig hos 19% av pasientene. Fibrose var konsentrert i de nedre delene av lungene og var ikke assosiert med pågående eller tidligere røyking. Pasienter med høygradig lungefibrose hadde lavere lungefunksjonstestverdier, kortere seks minutters gangavstand, høyere gjennomsnittlig NYHA funksjonsklasse og økt dødelighet. En nylig publisert retrospektiv kohortstudie av 39 MCTD-pasienter sammenlignet baseline og oppfølging av lungefunksjons og HRCT-data over en tiårsperiode (47). Ved baseline hadde 51% av pasientene unormale lungefunksjonstesting. Forsert vital kapasitet (FVC) ble svakt redusert ved baseline (77% av forventet), men forble stabil etter 10 år.
Pre-kapillær PH er en viktig årsak til sykelighet og dødelighet ved systemiske bindevevssykdommer spesielt ved SSc og MCTD (48). I henhold til de Europeiske retningslinjene fra 2015 fra European Society of Cardiology (ESC) og European Respiratory Society (ERS) (49), er pre-kapillær pulmonal hypertensjon definert som et gjennomsnittlig lungearterietrykk (mPAP) på 25 mm Hg eller mer i ro ved høyre side hjertekateterisering, sammenfallende med et lungeinnkiletrykk (PAWP) på 15 mm Hg eller mindre. Det er foreslått en ny definisjon på pulmonal hypertensjon som har en mer statistisk tilnærming. Frisk befolkning har mPAP på 14,0 ±3,3 mmHg og 97,5% av befolkning har mPAP < 20 mmHg. Det er derfor foreslått at mPAP >20 mmHg i ro foreslås som PH. Samtidig er det foreslått at lungekarmotstand (PVR) ⩾3 Wood med uendret definisjon på lungeinnkiletrykk (PAWP) ⩽15 mmHg (50).
Ifølge den internasjonale klassifiseringen av pulmonal hypertensjon fra 2013 (51), hører pasienter med systemisk bindevevssykdom og prekapillær PH til to hovedkategorier. Til de som har isolert pulmonal arteriell hypertensjon (PAH) (gruppe 1.4.1) eller PH assosiert med interstitial lungesykdom (PH-ILD) (gruppe 3.2).
Data om PH i MCTD er begrenset. Det nasjonale PAH-registeret i Storbritannia (med en befolkning på rundt 60 millioner) (52) inkluderte 36 MCTD-pasienter med PH, noe som indikerer en total MCTD-PH-prevalens på 0,6 per million.
Tre tidligere små oppfølgingsstudier med en enkelt senter kohort estimerte den totale frekvensen av PH i voksen MCTD til 23-24% (25, 26, 53). PH ser ut til å være hyppig (6-9%) i barne- og ungdoms kohort av MCTD (jMCTD) kohorter med ett senter (54, 55). I den norske kohortstudien ble alle MCTD-pasienter screenet ved studieregistrering ved ekkokardiografi, og pasienter som mistenkes å ha PH ble henvist til høyre hjertekateterisering. Etter påmelding ble pasientene fulgt gjennomsnittlig i 5,6 år. Ved baseline hadde 2,0% av pasientene (3/147) PH. To ytterligere PH-pasienter ble identifisert under oppfølgingen, noe som ga en total PH-frekvens i kohorten på 3,4% (5/147). To av disse pasientene hadde PAH, og tre hadde PH-ILD (56). En nylig publisert studie fra Ungarn rapporterte at 18% av MCTD-pasientene (50/280) utviklet PAH etter en gjennomsnittlig oppfølgingstid på 14,5 år (32). Det er flere mulige årsaker til mye høyere PH-frekvens observert i denne studien. Det mest åpenbare er forskjellen i oppfølgingstid. En annen mulig forskjell er pasientutvalget: Den norske studien var populasjonsbasert, mens den ungarske kohorten ble avledet fra et stort CTD-senter (7, 32).
Det er ingen publiserte CTD-PAH-epidemiologiske studier fra Asia, men nyere data fra en japansk kohortstudie med ett senter av 70 pasienter med PAH assosiert ved systemiske bindevevssykdommer (PAH-CTD) (57) viste at MCTD var som oftest assosieres med PAH (43%, 30/70), etterfulgt av SLE (29%, 20/70) og SSc (19%, 13/70). Dette i motsetning til studier fra Europa og USA der flertallet av pasientene med PAH-CTD har SSc. For eksempel i den nasjonale britiske registerundersøkelsen som ble referert over, hadde 8% MCTD (36/429) (52). I det amerikanske baserte REVEAL-prospektive PAH-registeret (register for å evaluere tidlig og langvarig PAH-administrasjon) hadde 9% MCTD (58).
Affeksjon av hud er inkludert i kriteriene Sharp og Kasukawa (3, 5). Det mangler data om utslett ved MCTD, men den langtidsstudie av Burdt og kolleger, beskrevet erythematøs hudutslett ved debut hos 13% og kumulativt hos 53% av MCTD pasientene (25). Ungarsk kohortstudie av Hajas og kolleger rapporterte at kumulative 36% av pasientene utviklet affeksjon av hud. Dette inkluderte lysfølsomhet, utslett i kinn (malart utslett), telangiektasi eller hypo- og hyperpigmentering (32). I studien av Sullivan og kolleger hadde 29% malart utslett og 41% hadde alopeci (26). Interessant nok viste en liten patologistudie inkludert åtte pasienter med MCTD at epidermal patologi etterlignet den av subakutt kutan lupus erythematosus (SCLE), men en samtidig vaskulopati parallell med den som ble sett i DM skilte den fra SCLE (95).
Sklerodaktyli. Definisjonen sklerodaktyli er sammensatt av det greske “skleros” som betyr hardt og “daktylos” som betyr finger eller tå. I klassifikasjonen for systemisk sklerose er det nødvendig å ha forandringer proksimalt til proksimal interfalangeal (PIP) ledd for å klassifisere det som sklerodactyli ved systemisk sklerose (SSc) (59).
Tilgjengelige data indikerer at øsofageal dysmotilitet er vanlig i MCTD (7, 32, 60, 61). Samlet sett ser det ut til at andre GI-komplikasjoner enn dysmotilitet av spiserøret er sjelden i MCTD. Det er isolerte rapporter om MCTD med autoimmun hepatitt (62-64), primær biliær kolangitt (PBC) (65), portalhypertensjon (66), og proteintapende enteropati (67, 68). Angiodysplasi i magesekk (GAVE/’watermelon stomach’) som er en anerkjent komplikasjon av SSc (69), er svært sjelden og kun rapportert hos en MCTD-pasient (70).
Hematologiske manifestasjoner
Det foreligger ingen nyere data om hematologiske endringer i MCTD, men eldre studier indikerer at anemi og leukopeni er vanlig i løpet av sykdomsforløpet, mens trombocytopeni rapporteres å være sjelden (7, 24, 25, 32).
Kranial nevropati i form av trigeminus nevropati er den eneste nevrologiske komplikasjonen som inngår i MCTD kriteriene (71). Sentralnervesystem komplikasjoner med nevropsykiatriske manifestasjoner som ofte kan sees ved SLE ser ut til å være svært sjeldent ved MCTD (72). En undersøkelse fra Ungarn viste at sensornevralt hørselstap var dobbelt så hyppig hos pasienter med MCTD sammenlignet med matchete friske kontroller (73), men man har ingen videre undersøkelser som har bekreftet dette.
Nyreaffeksjon er ikke inkludert i noen av de fire kriteriene for MCTD. Samlet data fra flere studier indikerer at opptil en femtedel av MCTD-pasienter etter hvert vil utvikle nyreaffeksjon, son oftest et immunkompleks nefropati og histologisk klassifisert som membranøs glomerulonefritt (34, 74). Det ser imidlertid ut til at nyremedvirkning ved MCTD stort sett er subklinisk, og har god prognose. I den norske MCTD kohort studie hadde mindre enn 3% mindre nyreaffeksjon i løpet av sykdommen (upubliserte data). I en ungarsk kohortstudie hadde 4% nyreaffeksjon. Av de med nyreaffeksjon hadde 27% (3/11) trombotisk trombocytopenisk purpura-assosiert nefropati, men de resterende 73% (8/11) hadde glomerulonefritt, de aller fleste ISN/RPS 2003 klasse II mesangial glomerulonefritt (7, 75).
Behandling av MCTD
Ingen randomiserte kontrollerte studier på behandling av MCTD er blitt utført. Valg av terapi ved MCTD er basert på kunnskap og erfaring fra behandling av andre relaterte systemiske bindevevssykdommer som SLE, SSc og inflammatorisk myositt (2). Det foreligger ingen responskriteria og ingen publiserte behandlingsretningslinjer verken nasjonalt, eller internasjonalt.
Iskemi i ekstremiteter. Symptomatisk behandling av Raynauds fenomen (RP), det vanligste symptomet på MCTD, består av generelle råd (holde i varmen, unngå skader, kutte ut røyking og redusere koffein bruk) (76). Behandling med orale kalsiumkanalblokkere reduserer total perifer motstand og øker perifer blodstrøm med effekter på RP (77). Nifedipin er ofte brukt, men andre dihydropyridin kalsiumkanalblokkere kan brukes, bl.a. amlodipin, felodipin, isradipin. Administrering av intravenøst prostaglandin, oftest iloprost, har dokumentert effekt på RP (76). Det er dokumentert en positiv effekt av nitroglyserin salva på RP, men for fleste andre intervensjoner er dokumentasjonene begrenset, motstridende eller ikke-eksisterende (22, 76). Behandlingen av iskemiske fingersår er utfordrende. Fra behandling av SSc vet vi at prostaglandin-infusjoner (78), fosfodiesterase 5-hemmeren sildenafil (79, 80) og digital sympatektomi (81, 82) har effekter i den akutte fasen. I tillegg ser det ut til at subkutane, lokale doser av Botulinum Toksin A kan være effektive (83, 84). Oral bruk av endotelin-1 reseptor-hemmeren, bosentan, reduserer risikoen for nye digitale sår (85-87), men i likhet med de andre medisinene er det ikke blitt evaluert ved MCTD.
Plaquenil. Det gamle anti-malaria medikamentet hydroksyklorokin (HCQ), er av mange ansett for å være den grunnleggende immunmodulerende behandlingen i MCTD, men i motsetning til SLE, er det ikke data som støtter denne oppfatningen (2).
Kortikosteroider og DMARDs. Hos pasienter med pleuritt, perikarditt, artritt, myositt og / eller interstitial lungesykdom (ILD), er per oral og/eller intravenøs kortikosteroider, fortsatt hjørnesteinen i terapien. Som ved behandling av andre systemiske bindevevssykdommer, brukes som oftest i kombinasjoner. Vanlige brukte andrelinje steroidbesparende behandlinger (DMARDs) i MCTD inkluderer; per oral eller sc. metotreksat, per oral azathioprin (AZA), mykofenolat mofetil (MMF), ciclosporin eller takrolimus. MCTD pasienter med alvorlige former av ILD kan også behandles med intravenøst evt. per oral cyklofosfamid (2).
Biologiske legemidler. Det er foreløpig liten erfaring med bruk av «biologisk» sykdomsmodifiserende medikamenter ved MCTD. Det er noen enkelte pasient-kasuistikker om vellykket behandling med B-celle deplesjons-terapi med rituksimab (88-94), hovedsakelig for behandlingsresistent artritt, myositt, trombocytopeni og ILD. Det er kun svært få rapporter om MCTD pasienter behandlet med tumor nekrose faktor alfa (TNF alfa) hemmere (95-97), muligens fordi de første rapportene indikerte alvorlige bivirkninger. Vi har likevel i vår egen sykehuspraksis noen få MCTD-pasienter med behandlingsresistent artritt som har blitt behandlet med vellykket anti-TNF alfa-behandling (upubliserte data) (2).
PAH-behandling ved MCTD. Det er ingen spesifikke retningslinjer for behandling av pulmonal hypertensjon (PH) assosiert med MCTD, og behandlingsalternativene er avledet fra studier av PAH-spesifikke terapier i idiopatisk PAH og PAH assosiert med SSc. Til slutt, i motsetning til SSc, er det rapporter om MCTD-pasienter med PAH som har svart på immunsuppressiv behandling alene (98). Hos de svært få pasienter med PAH som svarer på en vasodilatorisk behandling ved RHC (<5%), bør kalsiumkanalblokkere forsøkes (49). Pasienter med PAH assosiert til systemisk bindevevssykdom (CTD-PAH) blir noen ganger behandlet med antikoagulasjon, for å redusere risikoen for intrapulmonal trombose og tromboemboli, men antikoagulasjonsbehandlingen på denne indikasjonen er dårlig dokumentert (49, 99, 100). Pasienter med SSc-PAH har en høyere blødningsrisiko enn pasienter med idiopatisk PAH (99, 101, 102). Prospektive studier med undergruppeanalyse er nødvendige for å nøyaktig vurdere om økt blødningsrisiko oppveier den tilsynelatende fordelen med antikoagulasjon i disse pasientgruppene, men dette har vi ikke noen særlig informasjon om i de pasientene som har MCTD-PAH.
-Den mer spesifikke behandlingen av PAH består av per oral endotelin reseptorantagonister (ERA) (bosentan, ambrisentan eller macitentan), per oral fosfodiesterase 5-hemmere (sildenafil, tadalafil eller vardenafil) og subcutan eller intravenøs prostanoider (epoprostenol, treprostinil, ilioprost) og inhalert iloprost (49, 103-112). Nyere rapporter har vist at per oral selexipag, en selektiv prostacyklin-reseptor, medførte både isolert og i kombinasjon med annen PAH-behandling til betydelig redusert risikoen for død eller andre komplikasjon relatert til PH (113-115). Riociguat er en per oral guanylate cyclase (sGC) stimulator som gir økt produksjon av cGMP som er reseptor for nitrogenoksid (NO). Behandling av kronisk tromboembolisk pulmonal hypertensjon (CTEPH) er hovedindikasjonen. Det foreligger også dokumentasjon og indikasjon på behandling av PAH både som monoterapi eller i kombinasjon med endotelin-reseptorantagonist. Ved behandlingsresistente tilfeller kan kombinasjon av to til tre av disse behandlingsalternativene brukes. Det mest foretrukne terapeutiske alternativet er en oral behandling med endotelin reseptorantagonist (ERA) i kombinasjon med fosfodiesterase 5-hemmere (116). Intravenøs, inhalert eller subkutant administrering av prostanoid behandling har flere bivirkninger og alvorlige ulemper men er best dokumentert hos pasienter i NYHA/WHO funksjonsklasse IV (49).
Svangerskap
- Vennligst se info fra NKSR
Prognose ved MCTD
Skade. Vurderinger av utfall ved MCTD er sammensatt. Det foreligger ingen standardiserte utfalls- eller skademål for MCTD. Det er utfall i forhold til fenotypisk stabilitet av MCTD diagnosen. Det er også utfall assosiert til overlevelse og organskade.
Dødelighet. Kunnskap om dødelighet hos pasienter med MCTD er mangelfull og er basert på en håndfull små oppfølgingsstudier med kohorter bestående av 15-47 pasienter fra enkelsentra, med gjennomsnittlig observasjonstid mellom 5-17 år. Det foreligger to større kohortstudier med 147 og 280 pasienter. Dette utgjør til sammen ti studier (7, 25, 26, 32, 36, 56, 71, 117-120) som representerer totalt 644 MCTD pasienter hvorav knapt 10% hadde jMCTD. Disse studiene hadde totalt 77 dødsfall. Det er vanskelig å trekke meningsfylte konklusjoner fra disse studiene på grunn av heterogenitet og forskjellig oppfølgingstid og det faktum at det ikke var noen sammenligning med matchede kontroller fra den generelle populasjonen (2). Fem studier (25, 26, 32, 71, 119) med en tidsperiode lengre enn gjennomsnittet av 10 år (11-17 år) oppfølging, besto av totalt 406 pasienter og rapporterte 50 dødsfall som kumulativt ga en dødelighet på mellom 8 og 36% og gjennomsnittlig på rundt 12%, (25, 26, 32, 71, 119). Dødsårsakene i disse studiene var hovedsakelig relatert til PH og/eller ILD i tillegg til infeksjoner og malignitet. Hvis man ser isolert på de som hadde MCTD med debut i barns/ungdomsårene var dødelighetsraten rapportert til å være lavere, varierende mellom 2,8 og 7,9%, ifølge en landsomfattende japansk studie og en litteraturgjennomgang av Michels (54, 118). Pulmonal hypertensjon (PH) var viktig dødsårsak i en norske landsomfattende studie ansvarlig for totalt 25% (3/12) av alle dødsfall, etter en gjennomsnittlig oppfølging på 5,6 år (7, 56, 121). I en ungarsk regional kohortstudie hadde totalt 50 pasienter utviklet PAH etter gjennomsnittlig 14,5 år (± 3,7 år) fra diagnose. I tillegg ble PAH rapportert å være den viktigste dødsårsaken, og forårsaket 41% (9/22) av dødsfallene i kohorten (32).
World Health Organization (WHO) funksjonelle klasse (WHO-FC) fortsatt en av de viktigste prediktorene for overlevelse både ved diagnose og i oppfølging av PH (49, 122-124). En tidligere studie fra amerikanske REVEAL PAH registret viste en markant lavere tre-års overlevelse av pasienter med nylig diagnostisert SSc-assosiert PAH (51,2% ± 4,0%, n = 166) sammenlignet med pasienter med nylig diagnostisert annen (ikke-SSc) systemisk bindevevssykdom assosiert PAH, som også inkludert MCTD-tilfeller (76,4% ± 4,6%, n = 88) (58). En nylig studie fra en stor prospektiv PH-database fra Storbritannia hadde som mål å studere de kliniske og hemodynamiske egenskapene og overlevelsen for PAH-pasienter inkludert de med systemiske bindevevssykdommer (CTD-PAH). Studien identifiserte totalt 342 CTD-PAH-pasienter, hvorav 36 (11%) var anti-U1RNP-antistoff-positive (125). Pasienter med anti-U1-RNP antistoffer var gjennomsnittlig yngre og hadde mindre funksjonsnedsettelse enn de anti-U1RNP negative CTD-PAH pasientene. Dette slo også ut i en multivariabel analyse der ble anti-U1-RNP-positivitet assosiert med redusert dødelighet.
Interstitiell lungesykdom (ILD) ser ut til å være vanlig ved MCTD. ILD har innvirkning på lungefunksjon, generell fysisk kapasitet og dødelighet (121). For tiden er det få pålitelige oppfølgingsdata om ILD i MCTD og utilstrekkelige data om prognostiske faktorer for utvikling av ILD og for progresjon av ILD (47). Etter et gjennomsnitt på 4,2 år oppfølging i den norske kohortstudien, var dødeligheten hos pasienter med normal HRCT 3,3%, sammenlignet med 20,8% (p <0,01) hos pasienter med alvorlig lungefibrose (120). Nylig fant vi ut at anti-SSA/Ro52-antistoffer er en mulig markør for lungefibrose ved MCTD (126).
De aller fleste pasientene som har fått MCTD diagnose utvikler ikke en annen systemisk bindevevssykdom. Det har vært et dogma i revmatologien at mange har oppfattet MCTD som et steg i utvikling til andre systemisk bindevevssykdommer som; SLE, SSc eller PM/DM. I en oppfølging av den norske nasjonal MCTD kohorten med opprinnelig 147 pasienter var 11% (16/147) døde og kun 9,5% (14/147) utviklet annen systemisk bindevevssykdom. Der var 2,0% (3/14) som utviklet systemisk sklerose, 4,1% (6/14) utviklet SLE, 2,7% (4/14) revmatoid artritt og 0,7% (1/14) fikk diagnosen antisynthetase syndrom (7, 9).
- Sharp GC, Irvin WS, LaRoque RL, Velez C, Daly V, Kaiser AD, et al. Association of autoantibodies to different nuclear antigens with clinical patterns of rheumatic disease and responsiveness to therapy. Journal of Clinical Investigation. 1971 Feb;50(2):350-9. PubMed PMID: 4992992. PMCID: 291931.
- Gunnarsson R, Hetlevik SO, Lilleby V, Molberg O. Mixed connective tissue disease. Best practice & research Clinical rheumatology. 2016 Feb;30(1):95-111. PubMed PMID: 27421219.
- Kasukawa R, Tojo T, Miyawaki S, Yoshida H, Tanimoto K, Nobunaga M, et al. Prelimary Diagnostic Criteria for Classification of Mixed Connective Tissue Disease. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsevier Science Publishers B. V. (Biomedical Division); 1987. p. 41-7.
- Alarcón-Segovia D, Villarreal M. Classification and Diagnostic Criteria for Mixed Connective Tissue Disease. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsvier Science Publishers B.V. (Biomedical Division); 1987. p. 33-40.
- Sharp GC. Diagnostic Criteria for Classification of MCTD. In: Kasukawa R, Sharp GC, editors. Mixed connective tissue disease and anti-nuclear antibodies. Amsterdam: Elsevier Science Publishers B.V. (Biomedical Division); 1987. p. 23-30.
- Kahn MF, Appelboom T. Syndrome de Sharp. In: Kahn MF, Peltier AP, Mayer O, Piette JC, editors. Les maladies systémiques. 3rd. Paris: Flammarion; 1991. p. 545-56.
- Gunnarsson R, Molberg O, Gilboe IM, Gran JT, Group PS. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Annals of the Rheumatic Diseases. 2011 Jun;70(6):1047-51. PubMed PMID: 21398332.
- Ungprasert P, Crowson CS, Chowdhary VR, Ernste FC, Moder KG, Matteson EL. Epidemiology of Mixed Connective Tissue Disease, 1985-2014: A Population-Based Study. Arthritis care & research. 2016 Dec;68(12):1843-8. PubMed PMID: 26946215. PMCID: PMC5426802.
- Reiseter S, Gunnarsson R, Corander J, Haydon J, Lund MB, Aalokken TM, et al. Disease evolution in mixed connective tissue disease: results from a long-term nationwide prospective cohort study. Arthritis Res Ther. 2017 Dec 21;19(1):284. PubMed PMID: 29268795. PMCID: PMC5740892.
- Hetlevik SO, Flato B, Rygg M, Nordal EB, Brunborg C, Hetland H, et al. Long-term outcome in juvenile-onset mixed connective tissue disease: a nationwide Norwegian study. Ann Rheum Dis. 2016 Jun 9. PubMed PMID: 27283334.
- Flam ST, Gunnarsson R, Garen T, Norwegian MSG, Lie BA, Molberg O. The HLA profiles of mixed connective tissue disease differ distinctly from the profiles of clinically related connective tissue diseases. Rheumatology (Oxford). 2015 Mar;54(3):528-35. PubMed PMID: 25187641.
- Black CM, Maddison PJ, Welsh KI, Bernstein R, Woodrow JC, Pereira RS. HLA and immunoglobulin allotypes in mixed connective tissue disease. Arthritis and Rheumatism. 1988 Jan;31(1):131-4. PubMed PMID: 3345219.
- Ruuska P, Hameenkorpi R, Forsberg S, Julkunen H, Makitalo R, Ilonen J, et al. Differences in HLA antigens between patients with mixed connective tissue disease and systemic lupus erythematosus. Annals of the Rheumatic Diseases. 1992 Jan;51(1):52-5. PubMed PMID: ISI:A1992HC13500011.
- Kuwana M, Okano Y, Kaburaki J, Inoko H. Clinical correlations with HLA type in Japanese patients with connective tissue disease and anti–U1 small nuclear RNP antibodies. Arthritis & Rheumatism. 1996;39(6):938-42.
- Paradowska-Gorycka A, Stypińska B, Olesińska M, Felis-Giemza A, Mańczak M, Czuszynska Z, et al. Association of HLA-DRB1 alleles with susceptibility to mixed connective tissue disease in Polish patients. HLA. 2016;87(1):13-8.
- Greidinger EL, Hoffman RW. Autoantibodies in the pathogenesis of mixed connective tissue disease. Rheumatic Diseases Clinics of North America. 2005 Aug;31(3):437-50, vi. PubMed PMID: 16084317.
- Hoffman RW, Maldonado ME. Immune pathogenesis of Mixed Connective Tissue Disease: a short analytical review. Clinical immunology. 2008 Jul;128(1):8-17. PubMed PMID: 18439877.
- Nyman U, Lundberg I, Hedfors E, Pettersson I. Recombinant 70-Kd Protein Used for Determination of Autoantigenic Epitopes Recognized by Anti-Rnp Sera. Clinical and Experimental Immunology. 1990 Jul;81(1):52-8. PubMed PMID: ISI:A1990DL46100009.
- Hoffman RW, Sharp GC, Deutscher SL. Analysis of anti–u1 RNA antibodies in patients with connective tissue disease. Arthritis and Rheumatism. 1995;38(12):1837-44.
- Smolen JS, Klippel JH, Penner E, Reichlin M, Steinberg AD, Chused TM, et al. HLA-DR antigens in systemic lupus erythematosus: association with specificity of autoantibody responses to nuclear antigens. Ann Rheum Dis. 1987 Jun;46(6):457-62. PubMed PMID: 3498447. PMCID: Pmc1002164.
- Kattah NH, Kattah MG, Utz PJ. The U1-snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunological reviews. 2010 Jan;233(1):126-45. PubMed PMID: 20192997. PMCID: PMC3074261.
- Wigley FM. Clinical practice. Raynaud’s Phenomenon. New England Journal of Medicine. 2002 Sep 26;347(13):1001-8. PubMed PMID: 12324557.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheumatic Diseases Clinics of North America. 2005 Aug;31(3):465-81, vi. PubMed PMID: 16084319.
- Lundberg I, Hedfors E. Clinical course of patients with anti-RNP antibodies. A prospective study of 32 patients. The Journal of rheumatology. 1991 Oct;18(10):1511-9. PubMed PMID: 1765975.
- Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis and Rheumatism. 1999 May;42(5):899-909. PubMed PMID: 10323445.
- Sullivan WD, Hurst DJ, Harmon CE, Esther JH, Agia GA, Maltby JD, et al. A prospective evaluation emphasizing pulmonary involvement in patients with mixed connective tissue disease. Medicine (Baltimore). 1984;63(2):92-107.
- Maricq HR, LeRoy EC, D’Angelo WA, Medsger TA, Rodnan GP, Sharp GC, et al. Diagnostic potential of in vivo capillary microscopy in scleroderma and related disorders. Arthritis and Rheumatism. 1980;23(2):183-9.
- Pavlov-Dolijanovic S, Damjanov NS, Stojanovic RM, Vujasinovic Stupar NZ, Stanisavljevic DM. Scleroderma pattern of nailfold capillary changes as predictive value for the development of a connective tissue disease: a follow-up study of 3,029 patients with primary Raynaud’s phenomenon. Rheumatology International. 2011 Sep 8:1-7. PubMed PMID: 21901350.
- Mugii N, Hasegawa M, Matsushita T, Hamaguchi Y, Horie S, Yahata T, et al. Association between nail-fold capillary findings and disease activity in dermatomyositis. Rheumatology. 2011 June 1, 2011;50(6):1091-8.
- Ganczarczyk ML, Lee P, Armstrong SK. Nailfold capillary microscopy in polymyositis and dermatomyositis. Arthritis and Rheumatism. 1988;31(1):116-9.
- Olesinska M, Czuszynska Z, Felis-Giemza A, Walkiewicz-Pielaszek K, Więsik-Szewczyk E, Wierzba K, et al. Clinical course and nailfold capillaroscopy patterns of 69 patients with mixed connective tissue disease (MCTD). EULAR Berlin2012.
- Hajas A, Szodoray P, Nakken B, Gaal J, Zöld E, Laczik R, et al. Clinical Course, Prognosis, and Causes of Death in Mixed Connective Tissue Disease. The Journal of Rheumatology. 2013 May 1, 2013.
- Aringer M, Smolen JS. Mixed connective tissue disease: what is behind the curtain? BestPractResClinRheumatol. 2007;21(6):1037-49.
- Kitridou RC, Akmal M, Turkel SB, Ehresmann GR, Quismorio FP, Jr., Massry SG. Renal involvement in mixed connective tissue disease: a longitudinal clinicopathologic study. Seminars in Arthritis and Rheumatism. 1986 Nov;16(2):135-45. PubMed PMID: 3563525.
- Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease–an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). American Journal of Medicine. 1972 Feb;52(2):148-59. PubMed PMID: 4621694.
- Bennett RM, O’Connell DJ. Mixed connective tisssue disease: a clinicopathologic study of 20 cases. Seminars in Arthritis and Rheumatism. 1980;10(1):25-51.
- Lundberg I, Nennesmo I, Hedfors E. A clinical, serological, and histopathological study of myositis patients with and without anti-RNP antibodies. Seminars in Arthritis and Rheumatism. 1992 Oct;22(2):127-38. PubMed PMID: ISI:A1992JT72800006.
- Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001 Feb;60(2):116-23. PubMed PMID: 11156543. PMCID: PMC1753477.
- Pope JE. Other manifestations of mixed connective tissue disease. Rheumatic Diseases Clinics of North America. 2005 Aug;31(3):519-33, vii. PubMed PMID: 16084323.
- Udoff EJ, Genant HK, Kozin F, Ginsberg M. Mixed connective tissue disease: the spectrum of radiographic manifestations. Radiology. 1977 Sep;124(3):613-8. PubMed PMID: 302009.
- Tehranzadeh J, Ashikyan O, Dascalos J, Dennehey C. MRI of large intraosseous lesions in patients with inflammatory arthritis. AJR American journal of roentgenology. 2004 Nov;183(5):1453-63. PubMed PMID: 15505320.
- Aalokken TM, Lilleby V, Soyseth V, Mynarek G, Pripp AH, Johansen B, et al. Chest abnormalities in juvenile-onset mixed connective tissue disease: assessment with high-resolution computed tomography and pulmonary function tests. Acta radiologica. 2009 May;50(4):430-6. PubMed PMID: 19277918.
- Kozuka T, Johkoh T, Honda O, Mihara N, Koyama M, Tomiyama N, et al. Pulmonary involvement in mixed connective tissue disease: high-resolution CT findings in 41 patients. Journal of Thoracic Imaging. 2001 Apr;16(2):94-8. PubMed PMID: 11292211.
- Prakash UBS. Lungs in mixed connective tissue disease. Journal of Thoracic Imaging. 1992;7(2):55-61. PubMed PMID: 1992163942.
- Saito Y, Terada M, Takada T, Ishida T, Moriyama H, Ooi H, et al. Pulmonary involvement in mixed connective tissue disease: comparison with other collagen vascular diseases using high resolution CT. J Comput Assist Tomogr. 2002;26(3):349-57.
- Wiener-Kronish JP, Solinger AM, Warnock ML, Churg A, Ordonez N, Golden JA. Severe pulmonary involvement in mixed connective tissue disease. American Review of Respiratory Disease. 1981 Oct;124(4):499-503. PubMed PMID: 7294510.
- Kawano-Dourado L, Baldi BG, Kay FU, Dias OM, Gripp TE, Gomes PS, et al. Pulmonary involvement in long-term mixed connective tissue disease: functional trends and image findings after 10 years. Clin Exp Rheumatol. 2015 Mar-Apr;33(2):234-40. PubMed PMID: 25896472.
- Lefèvre G, Dauchet L, Hachulla E, Montani D, Sobanski V, Lambert M, et al. Survival and Prognostic Factors in Systemic Sclerosis–Associated Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Arthritis & Rheumatism. 2013;65(9):2412-23.
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119. PubMed PMID: 26320113.
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan;53(1). PubMed PMID: 30545968.
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol. 2013 12/24/;62(25, Supplement):D34-D41. PubMed PMID: 24355639.
- Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, et al. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179(2):151-7.
- Alpert MA, Goldberg SH, Singsen BH, Durham JB, Sharp GC, Ahmad M, et al. Cardiovascular manifestations of mixed connective tissue disease in adults. Circulation. 1983 Dec;68(6):1182-93. PubMed PMID: 6640871.
- Kotajima L, Aotsuka S, Sumiya M, Yokohari R, Tojo T, Kasukawa R. Clinical features of patients with juvenile onset mixed connective tissue disease: analysis of data collected in a nationwide collaborative study in Japan. The Journal of rheumatology. 1996;23(6):1088-94.
- Mier RJ, Shishov M, Higgins GC, Rennebohm RM, Wortmann DW, Jerath R, et al. Pediatric-onset mixed connective tissue disease. Rheumatic Diseases Clinics of North America. 2005;31(3):483-96, vii.
- Gunnarsson R, Andreassen AK, Molberg O, Lexberg AS, Time K, Dhainaut AS, et al. Prevalence of pulmonary hypertension in an unselected, mixed connective tissue disease cohort: results of a nationwide, Norwegian cross-sectional multicentre study and review of current literature. Rheumatology (Oxford). 2013 Jul;52(7):1208-13. PubMed PMID: 23407386.
- Shirai Y, Yasuoka H, Okano Y, Takeuchi T, Satoh T, Kuwana M. Clinical characteristics and survival of Japanese patients with connective tissue disease and pulmonary arterial hypertension: a single-centre cohort. Rheumatology (Oxford). 2012 Oct;51(10):1846-54. PubMed PMID: 22740623.
- Chung L, Farber HW, Benza R, Miller DP, Parsons L, Hassoun PM, et al. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest. 2014 Dec;146(6):1494-504. PubMed PMID: 24992469. PMCID: PMC4251613.
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013 Nov;65(11):2737-47. PubMed PMID: 24122180. PMCID: 3930146.
- Ling TC, Johnston BT. Esophageal investigations in connective tissue disease: which tests are most appropriate? Journal of Clinical Gastroenterology. 2001;32(1):33-6.
- Lapadula G, Muolo P, Semeraro F, Covelli M, Brindicci D, Cuccorese G, et al. Esophageal motility disorders in the rheumatic diseases: a review of 150 patients. Clinical and Experimental Rheumatology. 1994 Sep-Oct;12(5):515-21. PubMed PMID: 7842532.
- Matsumoto T, Kobayashi S, Shimizu H, Nakajima M, Watanabe S, Kitami N, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000 Oct;20(5):366-73. PubMed PMID: 11092254.
- Galvan VG, Oltra MR, Rueda D, Esteban MJ, Redon J. Severe acute hepatitis related to hydroxychloroquine in a woman with mixed connective tissue disease. Clinical Rheumatology. 2007 Jun;26(6):971-2. PubMed PMID: ISI:000246178800026.
- Inoue K, Okajima T, Tanaka E, Ando B, Takeshita M, Masuda A, et al. A case of Graves’ disease associated with autoimmune hepatitis and mixed connective tissue disease. Endocrine journal. 1999 Feb;46(1):173-7. PubMed PMID: 10426583.
- James O, Macklon AF, Watson AJ. Primary biliary cirrhosis–a revised clinical spectrum. Lancet. 1981 Jun 13;1(8233):1278-81. PubMed PMID: 6112603.
- Hirasaki S, Koide N, Ogawa H, Wada T, Sato A, Ujike K, et al. Mixed connective tissue disease associated with idiopathic portal hypertension and chronic thyroiditis. Journal of gastroenterology. 1997 Dec;32(6):808-11. PubMed PMID: 9430021.
- Furuya T, Suzuki T, Onoda N, Tamura K, Sato K, Demura H, et al. Mixed connective tissue disease associated with protein losing enteropathy: successful treatment with intravenous cyclophosphamide therapy. Internal Medicine. 1992 Dec;31(12):1359-62. PubMed PMID: 1300171.
- Chao CT, Lai CY, Chang PL, Wu HS, Wu VC, Wang WJ. Mixed connective tissue disease with protein-losing enteropathy: discussion of the treatment strategy. Journal of Clinical Rheumatology. 2011 Aug;17(5):286-7. PubMed PMID: 21778905.
- Hung EW, Mayes MD, Sharif R, Assassi S, Machicao VI, Hosing C, et al. Gastric antral vascular ectasia and its clinical correlates in patients with early diffuse systemic sclerosis in the SCOT trial. J Rheumatol. 2013 Apr;40(4):455-60. PubMed PMID: 23418384. PMCID: Pmc3652008.
- Liberski SM, McGarrity TJ, Hartle RJ, Varano V, Reynolds D. The watermelon stomach: long-term outcome in patients treated with Nd:YAG laser therapy. Gastrointestinal Endoscopy. 1994 Sep-Oct;40(5):584-7. PubMed PMID: 7988823.
- Nimelstein SH, Brody S, McShane D, Holman HR. Mixed connective tissue disease: a subsequent evaluation of the original 25 patients. Medicine. 1980 Jul;59(4):239-48. PubMed PMID: 6967141.
- Nowicka-Sauer K, Czuszynska Z, Majkowicz M, Smolenska Z, Jarmoszewicz K, Olesinska M, et al. Neuropsychological assessment in mixed connective tissue disease: comparison with systemic lupus erythematosus. Lupus. 2012 Aug;21(9):927-33. PubMed PMID: 22433916.
- Hajas A, Szodoray P, Barath S, Sipka S, Rezes S, Zeher M, et al. Sensorineural hearing loss in patients with mixed connective tissue disease: immunological markers and cytokine levels. The Journal of rheumatology. 2009 Sep;36(9):1930-6. PubMed PMID: 19684145.
- Kobayashi S, Nagase M, Kimura M, Ohyama K, Ikeya M, Honda N. Renal involvement in mixed connective tissue disease. Report of 5 cases. American Journal of Nephrology. 1985;5(4):282-9. PubMed PMID: 2931986.
- Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The Classification of Glomerulonephritis in Systemic Lupus Erythematosus Revisited. Journal of the American Society of Nephrology. 2004 February 1, 2004;15(2):241-50.
- Huisstede BM, Hoogvliet P, Paulis WD, van Middelkoop M, Hausman M, Coert JH, et al. Effectiveness of interventions for secondary Raynaud’s phenomenon: a systematic review. Archives of physical medicine and rehabilitation. 2011 Jul;92(7):1166-80. PubMed PMID: 21704799.
- Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology. 2005 Feb;44(2):145-50. PubMed PMID: 15546967.
- Pope J, Fenlon D, Thompson A, Shea B, Furst D, Wells George A, et al. Iloprost and cisaprost for Raynaud’s phenomenon in progressive systemic sclerosis. Cochrane Database of Systematic Reviews [Internet]. 1998; (2).
- Brueckner CS, Becker MO, Kroencke T, Huscher D, Scherer HU, Worm M, et al. Effect of sildenafil on digital ulcers in systemic sclerosis: analysis from a single centre pilot study. Annals of the Rheumatic Diseases. 2010 August 1, 2010;69(8):1475-8.
- Moinzadeh P, Hunzelmann N, Krieg T. Combination therapy with an endothelin-1 receptor antagonist (bosentan) and a phosphodiesterase V inhibitor (sildenafil) for the management of severe digital ulcerations in systemic sclerosis. J Am Acad Dermatol. 2011 Sep;65(3):e102-4. PubMed PMID: 21839301.
- Hartzell TL, Makhni EC, Sampson C. Long-term results of periarterial sympathectomy. The Journal of hand surgery. 2009 Oct;34(8):1454-60. PubMed PMID: 19683883.
- Wasserman A, Brahn E. Systemic sclerosis: Bilateral improvement of Raynaud’s phenomenon with unilateral digital sympathectomy. Seminars in Arthritis and Rheumatism. 2010;40(2):137-46.
- Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum. 2012 Feb;41(4):599-603. PubMed PMID: 21868066.
- Neumeister MW. The role of botulinum toxin in vasospastic disorders of the hand. Hand clinics. 2015 Feb;31(1):23-37. PubMed PMID: 25455354.
- Dhillon S. Bosentan a review of its use in the management of digital ulcers associated with systemic sclerosis. Drugs. 2009;69(14):2005-24. PubMed PMID: ISI:000270768600010.
- Korn JH, Mayes M, Matucci Cerinic M, Rainisio M, Pope J, Hachulla E, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis and Rheumatism. 2004 Dec;50(12):3985-93. PubMed PMID: 15593188.
- Tsifetaki N, Botzoris V, Alamanos Y, Argyriou E, Zioga A, Drosos AA. Bosentan for digital ulcers in patients with systemic sclerosis: a prospective 3-year followup study. The Journal of rheumatology. 2009 Jul;36(7):1550-2. PubMed PMID: 19567637.
- Dunkley L, Green M, Gough A. Comment on: a case of Raynaud’s phenomenon in mixed connective tissue disease responding to Rituximab therapy response. Rheumatology. 2007 Oct;46(10):1628-9. PubMed PMID: 17766999.
- Haroon M, O’Gradaigh D, Foley-Nolan D. A case of Raynaud’s phenomenon in mixed connective tissue disease responding to rituximab therapy. Rheumatology. 2007 Apr;46(4):718-9. PubMed PMID: 17289791.
- Rudolph SE, Kouba M, Hrdlicka P. [Severe corticoid-refractory autoimmune thrombocytopenia associated with mixed connective tissue disease (Sharp’s syndrome). Treatment with rituximab]. Deutsche Medizinische Wochenschrift. 2009 Sep;134(36):1734-8. PubMed PMID: 19718594.
- Vela P, Sivera F, Batlle-Gualda E, Mayor M, García-Manzanares A, Pascual E. Severe ischemia following treatment with rituximab in a patient with mixed connective tissue disease: an unusual complication. Lupus. 2010 July 1, 2010;19(8):1005-6.
- Gunnarsson R, Gilboe I-M, Garen T, Molberg Ø. [1928] Evaluating the Therapeutic Effects of B Cell Depletion Therapy with Rituximab in a Longitudinal Cohort of Mixed Connective Tissue Disease Patients. American College of Rheumatology The 76th 2012 Annual Scientific Meeting; Nov 9-14, 2012; Washington D.C. USA: Arthritis & Rheumatism; 2012. p. S1-S1216.
- Pascual M, Narváez J, Espi GA, Recalde MLd, Zacarias A, Alegre JJ. [277] Rituximab in Refractory Mixed Connective Tissue Disease: An Observational Study. American College of Rheumatology The 79th 2015 Annual Scientific Meeting; November 7-11, 2015; San Francisco, USA: Arthritis & Rheumatology; 2015.
- Jansson AF, Sengler C, Kuemmerle-Deschner J, Gruhn B, Kranz AB, Lehmann H, et al. B cell depletion for autoimmune diseases in paediatric patients. Clinical Rheumatology. 2011 Jan;30(1):87-97. PubMed PMID: 21120559.
- Richez C, Blanco P, Dumoulin C, Schaeverbeke T. Lupus erythematosus manifestations exacerbated by etanercept therapy in a patient with mixed connective tissue disease. Clinical and Experimental Rheumatology. 2005;23(2):273.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alpha agents. Seminars in Arthritis and Rheumatism. 2008;37(6):381-7.
- Christopher-Stine L, Wigley F. Tumor necrosis factor-alpha antagonists induce lupus-like syndrome in patients with scleroderma overlap/mixed connective tissue disease. The Journal of rheumatology. 2003;30(12):2725-7.
- Jais X, Launay D, Yaici A, Le Pavec J, Tcherakian C, Sitbon O, et al. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum. 2008 Feb;58(2):521-31. PubMed PMID: 18240255.
- Roldan T, Landzberg MJ, Deicicchi DJ, Atay JK, Waxman AB. Anticoagulation in patients with pulmonary arterial hypertension: An update on current knowledge. The Journal of Heart and Lung Transplantation. 2015 Oct 9. PubMed PMID: 26527532.
- Ezedunukwe IR, Enuh H, Nfonoyim J, Enuh CU. Anticoagulation therapy versus placebo for pulmonary hypertension. Cochrane Database Syst Rev. 2014 Jun 3;6:Cd010695. PubMed PMID: 24887213.
- Nikpour M, Stevens W, Proudman SM, Buchbinder R, Prior D, Zochling J, et al. Should patients with systemic sclerosis-related pulmonary arterial hypertension be anticoagulated? Internal Medicine Journal. 2013;43(5):599-603.
- Henkens IR, Hazenoot T, Boonstra A, Huisman MV, Vonk-Noordegraaf A. Major bleeding with vitamin K antagonist anticoagulants in pulmonary hypertension. Eur Respir J. 2013 Apr;41(4):872-8. PubMed PMID: 22936704.
- Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, et al. Bosentan Therapy for Pulmonary Arterial Hypertension. New England Journal of Medicine. 2002 Mar 21;346(12):896-903. PubMed PMID: 11907289.
- Galie N, Badesch D, Oudiz R, Simonneau G, McGoon MD, Keogh AM, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005 Aug 2;46(3):529-35. PubMed PMID: 16053970.
- Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. New England Journal of Medicine. 2005 Nov 17;353(20):2148-57. PubMed PMID: 16291984.
- Caravita S, Wu SC, Secchi MB, Dadone V, Bencini C, Pierini S. Long-term effects of intermittent Iloprost infusion on pulmonary arterial pressure in connective tissue disease. European journal of internal medicine. 2011 Oct;22(5):518-21. PubMed PMID: 21925064.
- Jing Z-C, Yu Z-X, Shen J-Y, Wu B-X, Xu K-F, Zhu X-Y, et al. Vardenafil in Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine. 2011 2011/06/15;183(12):1723-9. PubMed PMID: 21471085.
- Klinger JR. Tadalafil for the treatment of pulmonary arterial hypertension. Expert Review of Respiratory Medicine. 2011 2011/06/01;5(3):315-28.
- Galiè N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, et al. Updated Treatment Algorithm of Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2013 Dec 24;62(25_S):D60-72. PubMed PMID: 24355643.
- Granton J, Mercier O, De Perrot M. Management of Severe Pulmonary Arterial Hypertension. Semin Respir Crit Care Med. 2013 //13.09.2013;34(05):700-13. PubMed PMID: 101055S00331356460.
- Humbert M. Current Challenges in Pulmonary Hypertension. Semin Respir Crit Care Med. 2013 //13.09.2013;34(05):549-50. PubMed PMID: 101055S00331356495.
- Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani H-A, et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. New England Journal of Medicine. 2013 Aug 29;369(9):809-18. PubMed PMID: 23984728.
- Simonneau G, Torbicki A, Hoeper MM, Delcroix M, Karlócai K, Galiè N, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. European Respiratory Journal. 2012 October 1, 2012;40(4):874-80. PubMed PMID: 22362844.
- Sharma K. Selexipag for the Treatment of Pulmonary Arterial Hypertension. Expert Rev Respir Med. 2015 Nov 15. PubMed PMID: 26567613.
- Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. New England Journal of Medicine. 2015;373(26):2522-33. PubMed PMID: 26699168.
- Hassoun PM, Zamanian RT, Damico R, Lechtzin N, Khair R, Kolb TM, et al. Ambrisentan and Tadalafil Up-front Combination Therapy in Scleroderma-associated Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine. 2015 2015/11/01;192(9):1102-10.
- Grant KD, Adams LE, Hess EV. Mixed connective tissue disease – a subset with sequential clinical and laboratory features. The Journal of rheumatology. 1981 Jul-Aug;8(4):587-98. PubMed PMID: 6975376.
- Michels H. Course of mixed connective tissue disease in children. Annals of Medicine. 1997 Oct;29(5):359-64. PubMed PMID: 9453280.
- Piirainen HI. Patients with arthritis and anti-U1-RNP antibodies: a 10-year follow-up. British Journal of Rheumatology. 1990 Oct;29(5):345-8. PubMed PMID: 2224402.
- Singsen BH, Bernstein BH, Kornreich HK, King KK, Hanson V, Tan EM. Mixed connective tissue disease in childhood. A clinical and serologic survey. Journal of Pediatrics. 1977 Jun;90(6):893-900. PubMed PMID: 300795.
- Gunnarsson R, Aalokken TM, Molberg O, Lund MB, Mynarek GK, Lexberg AS, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Annals of the Rheumatic Diseases. 2012 Dec;71(12):1966-72. PubMed PMID: 22550317.
- Barst RJ, Chung L, Zamanian RT, Turner M, McGoon MD. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest. 2013 Jul;144(1):160-8. PubMed PMID: 23429998.
- Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012 Mar;39(3):589-96. PubMed PMID: 21885392.
- Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, et al. FIve-year outcomes of patients enrolled in the reveal registry. Chest. 2015;148(4):1043-54.
- Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, et al. Characteristics and survival of patients with anti-U1RNP antibodies in connective tissue disease associated pulmonary arterial hypertension. Arthritis & Rheumatology. 2015:n/a-n/a.
- Gunnarsson R, El-Hage F, Aaløkken TM, Reiseter S, Lund MB, Garen T, et al. Associations between anti-Ro52 antibodies and lung fibrosis in mixed connective tissue disease. Rheumatology (Oxford). 2015 August 28, 2015. PubMed PMID: 26320136.
