BINDEVEVSSYKDOMMER (REV 021-033)
56 Inklusjonslegeme myositt (IBM) (REV 021)
Inklusjonslegememyositt
Øyvind Palm and Jan Tore Gran
Kjennetegn på inklusjonslegeme myositt
Alder over 45 år ved debut, langsom sykdomsprogresjon, moderat forhøyet CK i blodet.
Muskelatrofi, også distalt i underarmer, hender og legger.
Manglende behandlingsrespons på kortikosteroider.
Anti-cN1A antistoff positiv hos ca. 40%, men er ikke spesifikt.
Diagnosen sikres ved muskelbiopsi (og elektronmikroskopi).
Prosedyrekoder: 6-minutter gangtest: FYFX05. EKG: FPFE15
Definisjon
Inklusjonslegememyositt (IBM) er en inflammatorisk muskelsykdom som skiller seg fra andre inflammatoriske myositter ved et langsommere sykdomsforløp og ofte asymmetrisk muskelaffeksjon distalt i ekstremitetene. Tilstanden rammer vanligvis eldre personer, og kortikosteroider har ingen effekt. Anti-cN1A-antistoff er positivt hos omtrent 40 % av pasientene, men er ikke spesifikt for IBM. Histologisk karakteriseres sykdommen av inklusjons-legemer, vakuoler og inflammasjon i muskel.
Selv om IBM er en inflammatorisk muskelsykdom, er inflammasjonen mindre fremtredende enn ved andre myositter som polymyositt og dermatomyositt, immunmediert nekrotiserende myopati (IMNM), antisyntetase syndromet og andre overlapp-syndromer med myositt (Roy B, 2023).
Historie
IBM ble først beskrevet som en egen sykdom i 1978 (Carpenter, S, 1978). Til sammenligning ble polymyositt beskrevet i 1887 (Unverricht, H. Polymyositis acuta progressiva. Z. Klin.Med. 12, 533–549, 1887) og dermatomyositt utskilt som subgruppe i 1891 (Uverricht, H. Dermatomyositis acuta. Dtsch. Med. Wochenschr. 17, 41–44, 1891). Sammenheng med
Patogenese
Autoimmunitet. I motsetning til tidligere oppfatninger, hvor IBM ble ansett som en primær nevrodegenerativ sykdom, tyder nyere funn på at autoimmunitet spiller en rolle. Antistoff mot et 43-kDa protein (cytosolisk 5′-nukleotidase 1A-antistoff/anti-cN1A) er påvist hos en andel av pasientene, men dette antistoffet er ikke spesifikt for IBM. Dessuten påvises antistoffet kanskje hos mindre enn halvparten av pasientene (Roy B, 2023).
Muskelbiopsier fra IBM-pasienter viser økt forekomst av biomarkører for T-celleautoimmunitet, inkludert CD8+ cytotoksiske T-celler, relaterte kemokiner og cytokiner, faktisk mer enn i andre former for myositt. I tillegg har hel-genom screening studier (GWAS) vist at IBM ikke skyldes en uidentifisert arvelig genmutasjon (Britson, K. A. 2018).
Vakuoler / inklusjonslegemer. Det sees en beskjeden grad av endomyseal inflammasjon hvor celleinfiltratet består av CD8+ cytotoksiske T-celler. Omkring 25 % av de mononukleære cellene som omgir eller invaderer non-nekrotiske fibre, består av makrofager, mens av de mononukleære cellene som invaderer nekrotiske fibre, utgjør makrofagene 80 %. Ragged-red fibres, som tyder på mitokondrieskade, sees i nesten alle muskelbiopsier. Vakuoler er ikke spesifikke for inklusjonslegeme myositt og kan påvises ved en rekke andre muskelsykdommer. Rimmed vacuoles betyr at vakuolen er begrenset av en tykk vegg (emerin og lamin NC) (Naddaf E, 2018). Filamenter sees i både cytoplasma og i nukleus. Disse tubulo-filamentose strukturene av membranøst cytoplasmatisk materiale opptrer som inklusjoner under lysmikroskop (vakuoler) og viser seg ved elektronmikroskopi å bestå av parede heliske filamenter, svært likt det man ser ved Alzheimers sykdom. Disse farges ved Kongo rødt, thioflavin og krystallfiolett som alle indikerer amyloidose (beta amyloid protein).Fiberatrofi av begge typer kan påvises.
. CC BY-NC 3.0")
Sporadiske og hereditære former
Inklusjonslegememyositt kan deles inn i sporadisk IBM (sIBM), familiær IBM (fIBM) og hereditær IBM (hIBM).
-Sporadisk inklusjonslegeme myositt. sIBM er den vanligste muskelsykdommen hos personer over 50 år (minimums insidens er 0,6 hos menn og 0,7 hos menn per år per 100 000). Den kan opptre med andre revmatiske sykdommer som systemisk sklerose, Sjøgrens syndrom, dermatomyositt, SLE og revmatoid artritt (de Paepe B, 2018).
-Hereditær (familiær/genetisk) IBM (hIBM). Den familiære formen er lik den sporadiske bortsett fra familiær forekomst (Ranque-Francois B, 2005). hIBM kalles også “Distal myopati med rimmed vacuoles” og er en genetisk sykdom. Denne debuterer blant tenåringer eller unge voksne (Broccolini A, 2014). I motsetning til sIBM sees det ved hIBM ingen positiv Kongo rød farging og ingen lymfocyttinfiltrasjon ved biopsi. hIBM er en heterogen gruppe som omfatter bl.a. autosomalt recessive former som hIBM hos persiske jøder, japansk hereditær distal myopati og autosomalt dominante former som Welanders distale myopati (svensker) og finsk tibial muskulær dystrofi. hIBM skal mistenkes hos yngre individer med progredierende distal muskelaffeksjon hvor quadriceps først affiseres sent i sykdomsforløpet (eller aldri). Som oftest autosomal recessiv (mutasjon av GNE- genet på kromosom 9), men finnes også i en sjelden autosomal dominant form.
For resten av dette kapitlet omtales imidlertid kun sporadisk inklusjonslegeme myositt, (sIBM).
Epidemiologi
sIBM rammer oftere menn enn kvinner (3 : 2) og debuterer vanligvis mellom 50 og 70 års alder (Molberg Ø, 2016). Den hereditære formen debuterer i 20-30 årene. Prevalens og insidens er sannsynligvis underestimert på grunn av feildiagnostisering. En norsk polulasjonsbasert studie fant relativt høy forekomst med prevalens 3,3 per 100.000 (Dobloug GC, 2014).
Symptomer
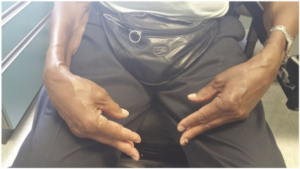
sIBM utvikler seg langsomt over flere år, vanligvis uten smerter. Gjennomsnittlig tid fra symptomstart til diagnose er 5-6 år (Dobloug GC, 2014). Sykdommen viser en jevn progresjon, men alvorlighetsgraden og prognosen varierer individuelt (Sangha G, 2021).
- Noen asymptomatiske, tidlige tilfeller påvises tilfeldig ved at muskelenzymet kreatin kinase (CK) er forhøyet i serum (Naddaf E, 2022).
Underekstremiteter. Sykdommen begynner nesten alltid i underekstremitetene, og det første symptomet kan være tendens til å falle fremover på grunn av svakhet i lårmusklene (vastus betydelig mer enn rectus femoris). Dermed er kne-ekstensjonen påvirket. Ofte ses asymmetri. Gangfunksjonen blir gradvis redusert, og det blir vanskeligere å gå i trapper og reise seg fra sittende stilling. Dersom gangfunksjonen skulle svikte, og ganghjelpemidler blir nødvendige innen ett år fra debut, bør en vurdere om en annen diagnose er mer sannsynlig (Naddaf E, 2022).
Distal muskulatur, som fingerbøyere og leggmuskler, affiseres ofte, i tillegg til proksimale muskler som lårmusklene. Typisk ses atrofi av volart underarm og svakhet i fingerbøyerne (spesielt flexor digitorum profundus), mens fingerekstensorene ofte spares. Pasienten håndgrep er ofte svakt.
Dysfagi forekommer hos 50-60 % av pasientene, men undersøkelser viser at forekomsten kan være høyere (se nedenfor). I noen tilfeller ses også crikofaryngeal sfinkter dysfunksjon. Dysfagi øker risikoen for aspirasjon og aspirasjonspneumoni. Nedsatt næringsinntak og vekttap kan også være et problem.
Svakhet i ansiktsmuskulatur kan forekomme rundt orbicularis oculi. Affeksjon av m. deltoideus forekommer nesten ikke. Ledsagende polynevropati er hyppig.
Drop-fot er observert hos 12% (Alamr M, 2022).
. CC BY 4.0")
Undersøkelser
IBM har klassiske kjennetegn, men krever en systematisk vurdering. Typisk svekkes muskler som kontrollerer finger-fleksjonen (gripeevnen), kne-ekstensjon og ankel dorsal-fleksjon. I motsetning til mange andre myopatier angripes i mindre grad muskler som styrer skulder- og hofte abduksjon, samt hofte-fleksjon. Ofte benevnes manifestasjonene som distal, selv om håndens egentlige distale muskler som m. interossei, tenar- og hypotenar vanligvis spares ved IBM.
Anamnesen kartlegger aktuelle symptomer (se ovenfor) med fokus på utvikling av svikt i distal og proksimal muskelkraft og atrofier, samt svelgeproblemer (øsofagus-funksjon).
Klinisk undersøkelse inkluderer generell status med observasjon av ev. muskelatrofi distalt i ekstremiteter og proksimal muskulatur. Hender vurderes for ev. fleksjonskontrakturer. En enkel kraft-test viser om pasienten reiser seg greit fra stol eller huk-sittende uten støtte og kan løfte armene lett over hodet. Svakhet i nakkemuskulatur kan etterprøves ved å gi motstand.
6-minutter gangtest gir en vurdering av fysisk gangfunksjon (antall meter tilbakelagt i løpet av 6 minutter rask gange). Ved kontroller over tid kan eventuell sykdomsprogresjon synliggjøres.
–Fysioterapeut. Standardiserte tester for muskelstyrke kan være viktige verktøy i oppfølging av pasienter, særlig for å vurdere ev sykdomsutvikling over tid eller om behandlingsmål oppnås. Fysioterapeuter tester både direkte muskelstyrke og utholdenhet i form av manuell og kvantitativ muskeltesting og isometriske muskeltester (Oldroyd AGS, 2020).
Laboratorieprøver kan omfatte inflammasjonsmarkører (CRP, SR), leukocytter med differensialtellinger, trombocytter, elektrolytter, kreatin kinase (CK), lever-, nyre-, og thyreoidea-funksjonsprøver, glukose og urin-stiks. SR oftest normal. CK forøket til 2-5 ganger øvre normal verdi. Antistoff (Anti-cN1A) er beskrevet nedenfor.
-Immunologiske undersøkelser: Ingen klassiske antistoffer forventes å foreligge. Anti-cytosolic 5′-nucleotidase 1A (cN-1A) har i en studie vist sensitivitet (evne til å fange opp IBM) på 50% (Felice KJ, 2018), men i andre kohorter noe lavere, ca. 30%. Antistoffet er dessverre ikke spesifikt for IBM og ses ved Sjøgrens syndrom (36%) (Salajegheh M, 2011) og systemisk lupus (20%) (Halilu F, 2022), selv uten muskelmanifestasjoner (Lloyd TE, 2016). cN-1A er tilgjengelig for analyse (blot) blant myositt-spesifikke antistoff. Økt cN-1A antistoff rapporteres å være 33 – 76% sensitiv og 92 -96% spesifikk for IBM (Pluk H, 2013; Larman HD, 2012).
Elektrofysiologiske undersøkelser. EMG og nevrografi kan brukes for å utelukke andre diagnoser som motornevronsykdom eller nevropati som kan ha lignende debutsymptomer. Siden MR og annen diagnostikk er mer brukt gjøres de elektrofysiologiske undersøkelsene nå sjeldnere enn tidligere (Lotz BP, 1989).
Bildediagnostikk
. CC BY-3.0")
MR av muskulatur kan bidra til å skille mellom IBM fra muskel-dystrofi. Hvis forandringene bare sees ved bruk av T2-STIR, tyder dette på IBM – ikke dystrofi. Typiske funn ved IBM er fettinfiltrasjon og atrofi av spesifikke muskelgrupper. De hyppigst affiserte muskelgruppene er fleksor digitorum profundus, anteriore muskler i proksimale underekstremiteter og mediale deler av gastrocnemius på leggen. Hos pasienter med påfallende atrofi av m. quadriceps, men klinisk bare litt redusert kne-ekstensjon, kan forklaringen være at m. rectus femoris spares ofte i tidlige sykdomsfaser. Fettinfiltrasjon og atrofi av vastus intermedius og vastus medialis-muskler med økende funn lengst distalt er typisk og ses hos en del av pasientene (Tasca G, 2015). Muskelvolumet kan dessuten si noe om sykdomsutviklingen (Roy B, 2023).
Ultralydundersøkelse av muskulatur kan vise samme fordeling av atrofi som ved MR.
Røntgen øsofagus. Barium kontrastmiddel ved dynamisk røntgen-undersøkelse vil avsløre at dysfunksjonen først og fremst rammer øvre del av spiserøret. Selv om bare omkring 50% merker svelgeproblemer, kan redusert øsofagus funksjon og risiko for aspirasjon påvises hos 86,5% (Alamr M, 2022).
Muskelbiopsi er avgjørende for å bekrefte diagnosen. Typiske histologiske funn er vakuoler, endomysial inflammasjon, intracellulære amyloidavleiringer og tubulofilamenter. Diagnostiske kriterier er utarbeidet for å sikre korrekt diagnose (se nedenfor). Vær oppmerksom på at IBM ikke er den eneste tilstanden som presentere “rimmed vacuoles” i biopsi. Ved klinisk mistanke må genetiske ikke-inflammatoriske tilstander (hIBM) utelukkes.
Typiske histologiske funn er :
- Vakuoler (“rimmed vacuoles”, “inclusion bodies”)
- Endomyseal inflammasjon
- Intracellulære amyloid-depoter
- Tubulofilamenter
Diagnostiske kriterier
| Klinikk og laboratorium | Klassifikasjon | Patologiske funn |
|---|---|---|
| Varighet > 12 måneder
Alder ved debut > 45år Kne-ekstensjon svakere enn hofte-fleksjon og/eller finger fleksjon svakhet > skulder abduksjon svakhet CK ikke høyere enn 15× øvre referanse-grense |
Klinikk‐patologisk definert IBM | Alle de følgende:
Endomyseal inflammatoriske infiltrater Rimmed vacuoles (inklusjonslegemer) Protein akkumulering a eller 15‐ to 18‐nm filamenter |
| Varighet > 12 måneder
Alder ved debut > 45år Kne-ekstensjon svakere enn hofte-fleksjon og/eller finger fleksjon svakhet > skulder abduksjon svakhet CK ikke høyere enn 15× øvre referanse-grense |
Sannsynlig / mulig IBM |
En eller flere, men ikke alle av de følgende: Endomyseal inflammatoriske infiltrater Rimmed vacuoles (inklusjonslegemer) Protein akkumulering a eller 15‐ to 18‐nm filamenter
|
a amyloid eller andre protein akkumuleringer ved etablert metode (for eksempel for amyloid Kongo rødt, krystall fiolett, thioflavin T/S, for andre proteiner p62, SMI‐31, TDP‐43).
Differensialdiagnoser
Felles for disse tilstandene er at de kan presentere seg med muskelsvakhet, som er et sentralt trekk ved IBM. Grundig klinisk undersøkelse, blodprøver (CK, autoantistoffer), EMG, bildediagnostikk (MR) og eventuelt muskelbiopsi er nødvendig for å skille mellom disse tilstandene og stille riktig diagnose.
- Amyloidose: Opphopning av amyloid protein i vev, inkludert muskler, kan gi muskelsvakhet og atrofi, som ved IBM, men ofte med systemiske manifestasjoner som affeksjon av hjerte, nyrer eller nervesystem.
- Annen myositt (f.eks. polymyositt, dermatomyositt): Inflammatoriske muskelsykdommer kan gi muskelsvakhet og forhøyede muskelenzymer, som ved IBM, men har ofte ulik fordeling av muskelsvakhet, respons på behandling og histopatologiske funn.
- Immunmediert nekrotiserende myopati (IMNM):
Begge gir muskelsvakhet og forhøyede muskelenzymer. IMNM har ofte raskere progresjon, bedre respons på immunsuppressiv behandling, og histopatologi viser nekrose av muskelfibre uten inklusjonslegemer. - Antisyntetasesyndrom (ASS): Kan gi muskelsvakhet og interstitiell lungesykdom. ASS har ofte systemiske manifestasjoner som feber, Raynauds fenomen, artritt, og spesifikke autoantistoffer (anti-Jo-1), i motsetning til IBM.
- Immunmediert nekrotiserende myopati (IMNM):
- Depresjon: Kan gi tretthet og redusert fysisk aktivitet, som kan mistolkes som muskelsvakhet.
- Medikamentinduserte myopatier: statiner (myalgi, svakhet, sjelden rabdomyolyse), kortikosteroider (muskelatrofi og katabolisme), kolkisin (muskelsvakhet og nevropati)., antipsykotika (nevroleptisk malignt syndrom med muskelstivhet, feber og rabdomyolyse), antivirale medikamenter (muskelsvakhet, myalgi, laktatacidose), hydroksyklorokin (myopati), amiodaron (myopati med muskelsvakhet og forhøyede muskelenzymer). Medikamentinduserte myopatier bedres ofte ved seponering.
- Myopatier forårsaket av endokrine sykdommer (hypothyreose, Cushings syndrom): Kan gi muskelsvakhet og tretthet. Endokrine myopatier er assosiert med spesifikke hormonelle forstyrrelser og har ofte andre symptomer relatert til disse.
- Nevrologisk sykdom, inklusiv motornevronsykdom/amyotrofisk lateralsklerose og polynevropati: Nevrologiske sykdommer kan gi muskelsvakhet og atrofi, som ved IBM, men involverer ofte øvre motornevroner, har ingen muskelømhet eller forhøyede muskelenzymer, og viser karakteristiske nevrofysiologiske funn.
- Non-inflammatorisk myopati/arvelige myopatier: (f.eks. muskeldystrofier, metabolsk myopati): Disse tilstandene kan gi progressiv muskelsvakhet og atrofi, som ved IBM, men har ofte en genetisk årsak, karakteristiske histopatologiske funn, og mangler betennelsesforandringer i muskulaturen. Arvelige myopatier har ofte en positiv familiehistorie og kan bekreftes med genetisk testing.
- Sarkoidose (med myopati): En granulomatøs sykdom som kan affisere muskler og gi muskelsvakhet, som ved IBM, men har ofte systemiske manifestasjoner som lungeinvolvering, hudforandringer og lymfadenopati.
- Spinal stenose: Kan gi muskelsvakhet og smerter i bena, spesielt ved gange.
Behandling
Standardbehandling for IBM er non-farmakologiske tiltak som fysioterapi, ergoterapi, egentrening, pasientinformasjon for å redusere risiko for fall og skader og psykologisk støtte. Ved dysfagi kan faryngoøsofagal dilatasjon eller cricofaryngeal myotomi vurderes (Oh TH, 2008).
Immundempende behandling har vanligvis ingen effekt ved IBM, og det er derfor ikke rutine å forskrive kortikosteroider, metotreksat eller immunglobuliner (Glaubitz S, 2020; Schmidt K, 2017). Det gjelder også blant pasienter med cN-1A antistoff. En observasjons-studie viste at behandlede pasienter kom heller dårligere ut enn de ubehandlede (Benveniste O, 2011). Det er derfor ikke rutine å forskrive immunsuppressiv medikasjon som kortikosteroider, metotreksat eller immunglobuliner. Selv om CK i blodprøver faller under medikasjon, er en eventuell klinisk effekt vanligvis kortvarig (Naddaf E, 2018). For å unngå unødvendig og potensielt skadelig medikasjon er viktig å avgrense inklusjonslegememyositt fra andre typer myositt.
Kortikosteroider. Generelt medfører prednisolon og andre steroider mer skade av bivirkninger enn nytte ved inklusjonslegeme myositt. Med tanke på at enkelte kan ha noe responds på prednisolon forsøkes likevel behandlingen i blant. Eventuell utprøving bør være i en begrenset periode for å se om konkrete behandlingsmål som bedret kraft i standardiserte målinger, mestring av konkrete fysiske oppgaver og lignende kan nås (Naddaf E, 2022).
csDMARDs som metotreksat (Badrising UA, 2002), azathioprin og mykofenolat er prøvd ut i flere studier. Dessverre har de i beste fall bare en forbigående effekt. Generelt brukes derfor ikke disse medikamentene (Naddaf E, 2022).
Immunglobuliner/ IVIg. Noen kan ha forbigående effekt av i.v. gammaglobulin IVIg (Oktagam), særlig overfor dysfagi (Dobloug C, 2012). Enkelte rapporterer også om bedre muskelstyrke. Imidlertid ser immunglobuliner ikke ut til å ha vedvarende effekt over lengre tid og er ikke vist å bremse sykdomsutviklingen. Behandlingen er ikke bivirkningsfri og derfor ikke generelt anbefalt, men prøves i enkelte tilfeller med uttalte svelgevansker (Naddaf E, 2022; Glaubitz S, 2020). Det pågår utprøving av nye legemidler for IBM (Naddaf E, 2018).
Biologiske legemidler. TNF-hemmere, anakinra, β-interferon-1a og andre er utprøvd, men uten nytteverdi til nå (Naddaf E, 2022).
Oppfølging
Regelmessig oppfølging er viktig, med fokus på svelgefunksjon, lungefunksjon, muskelsvakhet og fysisk funksjon (Naddaf E, 2022).
Prognose
IBM er en progressiv sykdom som ofte fører til betydelig funksjonsnedsettelse på sikt. Noen deler forløpet inn i tre varianter: raskt progredierende type, fokal asymmetrisk og sIBM assosiert med systemisk bindevevssykdom (best utfall). Prognosen varierer, men gjennomsnittlig klarer pasientene seg uten ganghjelpemidler i 7,5-10 år etter symptomdebut. Deretter brukes for eksempel staver eller stokk og etter 13-15 år rullestol (Greenberg SA, 2019).
Retningslinjer, anbefalinger og prosedyrer
Lundberg IE, 2017: EULAR Klassifikasjon av myositt
Revmatologisk forening/Legeforeningen 2020
