BARN MED REVMATISK SYKDOM. BARNEREVMATOLOGI (REV 053-062)
121 Juvenil dermatomyositt, JDM (REV 021, REV 054)
Dermatomyositt hos barn (JDM)
Øyvind Palm
Kjennetegn på juvenil dermatomyositt
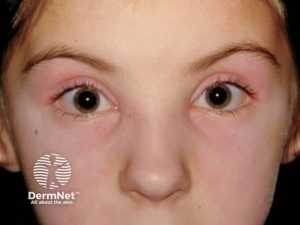
Ødematøst ansikt, særlig periorbitalt
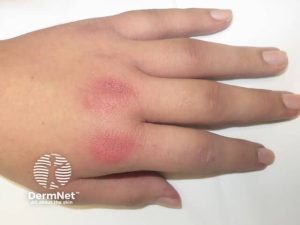
Eksantem: heliotropt, Gottrons, sjal, V-tegn. Kalsinose
Muskelsvakhet i underekstremiteter (gangvansker)
Blodprøver med forhøyet CK, myositt-spesifikke antistoff
MR lår viser muskelødem, biopsi bekrefter dermatomyositt
Polymyositt (uten hud-affeksjon) er uvanlig hos barn
ICD-10: M33.0; J99.1 (lunge-manifestasjon)
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A.
Definisjon
Juvenil dermatomyositt (JDM) er en autoimmun bindevevssykdom som angriper hud og muskler hos barn (18 år eller yngre). Kalsinose er også vanlig. I barnealder er det den vanligst formen for myositt. Sykdomsbildet er svært varierende og kan deles inn i subgrupper, basert på sykdomsforløp og antistoffprofil (se tabell nedenfor). Til forskjell fra dermatomyositt hos voksne er juvenil type ikke assosiert med maligne sykdommer (Meyer A, 2015; Li D, 2019).
Epidemiologi og Genetikk
JDM er svært sjelden med insidens på 2-4 tilfeller per million barn per år, men er likevel den vanligste inflammatoriske muskelsykdommen i barneårene (Meyer A, 2015).
JDM er en kompleks tilstand, også genetisk, der flere gener i HLA-regionen er involvert i sykdomsutviklingen (Miller FW, 2013).
Patogenese
Det antas at dendrittiske celler aktiveres av virale komponenter fra cellekjerner eller av vårt eget DNA. Dette utløser store mengder av type I interferoner (IFNα og IFNβ) som igjen aktiverer immunceller og fører til vaskulopati (Lee PY, 2007).
Symptomer
JDM er en heterogen sykdomsgruppe med ulike kliniske kjennetegn. Fenotype og forløp kan også bestemmes også være avhengig av antistoffmønsteret (Li D, 2019).
Hud: Dermatomyositt (med hud-symptomer) er vanligere enn polymyositt (uten hudsymptomer) hos barn. Hudmanifestasjonene omfatter:
- Heliotropt eksantem (lilla misfarging over øyne)
- Gottrons tegn (lilla over MCP, albuer, knær) og Gottrons papler (lilla knuter).
- V-tegn og Sjal-tegn er mindre vanlig enn hos voksne med dermatomyositt.
Anti-TIFγ-antistoffer disponere for alvorlig hud-manifestasjoner. Anti-Mi2-antistoffer er også assosiert med hudsykdom, men i liten grad med annen organmanifestasjon.
- Ødem. Ved sykdomsdebut kan huden være ødematøs, spesielt rundt øynene. Ved debut hos spebarn eller små barn med ødem på hender, bør man tenke på CANDLE syndrom som differensialdiagnose.
- Hypertrichose (økt hårvekst) er også beskrevet (Kayani M, 2023).
- Kalsinose under huden og langs muskler oppstår hos mange pasienter og kan utvikle seg til å bli hovedmanifestasjonen. Kalsinose oppstår vanligvis omtrent tre år fra sykdomsdebut og langvarig sykdom, lav alder ved sykdomsdebut og anti-NXP2-antistoffer er blant risikofaktorene (Balin SJ, 2012).
Muskler: Ikke alle med dermatomyositt har muskelaffeksjon (amyopatisk dermatomyositt), og myositt uten hudsykdom (polymyositt) er sjelden hos barn. Ved muskelsymptomer foreligger ofte symmetrisk, progredierende muskelsvakhet som kan objektiveres. Underekstremiteter rammes oftere enn overekstremiteter. Pasienter med anti-NXP2-antistoffer kan få alvorlig myositt. Anti-SRP- og anti-HMGCR-antistoffer kan tye på immunmediert nekrotiserende myositt som har et annet forløp enn klassisk polymyositt (Li D, 2019).
Hjertemuskelen kan også angripes. Ved uttalt muskelaffeksjon bør hjertefunksjon- og -rytme vurderes. Troponin forventes å stige med myokarditt (Mondal S, 2022).
Svelgefunksjon: Dysfagi og/eller dysmotilitet i øsofagus medfører redusert svelgefunksjon, som også kan sees hos voksne med myositt (Ebert EC, 2010).
Lungemanifestasjoner sees typisk ved antisyntetase syndrom, der en kombinasjon av interstitiell lungesykdom (ILD) og anti-Jo-1 eller ander typiske antistoffer (anti-PL7, anti-PL12, anti-OJ, anti-KS, anti-EJ, anti-Zo, anti-Ha) er til steede. MDA5-antistoffer disponerer også for lungemanifestasjoner (Palmucci S, 2022).
Utredning
Anamnesen kartlegger typiske symptomer i hud, muskler og andre organer (se ovenfor), samt feber, vekttap og generell helsetilstand.
Klinisk gjøre en generell statusundersøkelse med fokus på hjerte, lunger, blodtrykk, hud og muskler.
Laboratorieprøver. Inflammasjonsparametere er ikke alltid forhøyet, men kreatinkinase (CK), LD og ASAT er vanligvis moderat forhøyet. ALAT kan også stige litt. Noen pasienter utvikler myositt etter hudmanifestasjonene. CRP, SR, elektrolytter, nyrefunksjonsprøver, celletellinger og prøver for infeksjonsscreening anbefales for å utelukke tegn til andre diagnoser (Enders FB, 2017).
-Immunologiske prøver. Totalt har 60–95% av pasientene positive antistoffprøver. Antistoffmønsteret kan, sammen med det histologiske bildet, bidra til å karakterisere subgrupper av sykdommen og dermed være av behandlingsmessig og prognostisk betydning. Se tabellen nedenfor for en oversikt over kliniske undergrupper og relaterte antistoffer.
| Juvenil dermatomyositt med kliniske subgrupper og relaterte antistoff i blodet (Li D, 2019) | ||
| Kliniske subgrupper | Typiske antistoff (andel) | Kliniske kjennetegn |
| Dermatomyositt | ||
| -Hud og muskel-manifestasjon. Godt karakterisert ved muskelhistologi. Klinisk prognose og behandlingsrespons på behandlingen er variabel. | Anti-TIFγ (18-32%) | Alvorlig hud-sykdom, ulcerasjoner, lipodystrofi,. Muskelsykdommen kan være mild. Ofte et kronisk forløp. |
| Anti-NXP2 (1-23%) | Alvorlig muskelsykdom, gastrointestinal blødning og kalsinose. Remisjonsraten er lav. | |
|
Anti-MDA5 (7-38%) |
Artritt, ulcerasjoner. Raskt progredierende lunge-manifestasjoner (ILD). Økt mortalitet er påvist i Øst-Asiatisk kohorte. | |
| Anti-Mi2 (4-10%) | Lav risiko for organ-manifestasjoner eller alvorlig myositt. Responderer godt på behandling. | |
| Amyopatisk dermatomyositt | ||
| -Typiske dermatomyositt hud-manifestasjoner uten muskel-affeksjon. |
Anti-MDA5 (7–38%) |
Se Anti-MDA5 ovenfor |
| -Minimal eller progressive myositt er mer vanlig enn fravær av muskelsykdom |
Anti-TIF1γ (18–32%) |
Se Anti-TIF1γ ovenfor |
|
Anti-SAE (1%)
|
Muskelsykdommen kan utvikle seg senere i forløpet | |
| Immun-mediert nekrotiserende myositt | ||
| -Alvorlig muskelsykdom der histologi viser nekrose i myofibre, men med minimale inflammatoriske celleinfiltrater. Utslett er rapportert, men kan være atypisk. |
Anti-SRP (2%) |
Alvorlig myositt. Hjertemuskel-affeksjon. Behandlingsresistent. |
|
Anti-HMGCR (1%) |
Alvorlig muskelsykdom. Behandlingsresistens. | |
| Antisyntetase syndrom | ||
| -Myositt, ILD, Raynauds fenomen, artritt, feber og mekaniker hender |
Anti-Jo-1, anti-PL7, anti-PL12, anti-OJ, anti-KS, anti-EJ, anti-Zo, anti-Ha (til sammen <5%)
|
De ulike antisyntetase autoantistoffene er assosiert med muskel-dominant eller lunge-dominant sykdom hos voksne. For juvenil sykdom mangler data. |
| Overlappsykdom | ||
| -Pasienter fyller klassifikasjonskriterier for myositt, men i tillegg også for en annen revmatisk sykdom. Vanlig overlapp er systemisk sklerose, artritt og systemisk lupus (JSLE) |
Anti-PmScl (4–5%) Anti-U1RNP (4–6%) Anti-Ku, anti-U3RNP og anti-Scl70 |
Som primær-diagnose velges ut i fra de kliniske sykdomstrekk som dominerer. |
. CC BY-NC 4.0")
Bildediagnostikk. Ulike bildediagnostiske metoder er aktuelle vd utredning av JDM.
- Røntgen. Kalsinose kan identifiseres ved vanlig røntgen.
- MR er den foretrukne metoden for å påvise myositt, vanligvis i lårmusklene (Enders FB, 2017).
- Dynamisk røntgen av spiserøret eller manometri: Kan avdekke eventuell affeksjon av øsofagus
- HRCT (Høyoppløselig CT) av lunger kan brukes for å vurdere ev. lungemanifestasjoner (Enders FB, 2017).
Muskelbiopsi. I typiske tilfeller der diagnosen JDM er klar, er det ikke nødvendig med biopsi. Ved uklare eller atypiske tilfeller anbefales imidlertid biopsi for å bekrefte diagnosen (Enders FB, 2017).
Biopsien kan vise generelle tegn på muskelskader som degenerasjon, regenerasjon, nekrose og fagocytose, samt interstitielle ansamlinger av hvite blodceller. Ved dermatomyositt, som er den vanligste formen hos barn, ses også perimysial infiltrasjon (betennelse i bindevevet rundt muskelfibrene). Immunmediert nekrotiserende myopati viser nekrose i muskelfibrene, men med minimal betennelse. Polymyositt med endomysial infiltrasjon (betennelse i bindevevet mellom muskelfibrene) er sjelden hos barn.
Klassifikasjonskriterier
. CC BY NC-4.0")
EULAR/ACR-klassifikasjon for JDM og myositt er de samme som for voksne og inkluderer undergrupper basert på kliniske manifestasjoner og antistoffprofil (Lundberg E, 2017).
Differensialdiagnoser
Nedenfor er bare noen av de mulige differensialdiagnosene ved JDM. En grundig klinisk undersøkelse og supplerende undersøkelser, som blodprøver, MR og muskelbiopsi, er nødvendig for å stille riktig diagnose.
- Andre autoimmune sykdommer:
- Juvenil systemisk lupus erythematosus (jSLE): SLE kan gi hudutslett og muskelsmerter, men har ofte et bredere spekter av symptomer som involverer ledd, nyrer og andre organer.
- Juvenil idiopatisk artritt (JIA): Noen former for JIA kan gi muskelsvakhet og utslett, men artritt er vanligvis et mer fremtredende symptom.
- Medfødte muskelsykdommer:
- Muskeldystrofier: Disse sykdommene forårsaker progressiv muskelsvakhet og økt CK, men har vanligvis ikke hudutslett.
- Infeksjoner:
- Virusinfeksjoner/infeksiøs myositt: Noen virusinfeksjoner kan gi muskelsmerter og utslett, men disse symptomene er vanligvis forbigående. Virus (influenza B>A, enterovirus, EBV, SARS-CoV2). Påvirket allmenntilstand, CK-forhøyelse mulig (rabdomyolyse hos 3%), spontan tilbakegang (med normalisering av CK) etter 1-30 dager. Bakteriell myositt er sjelden (S. aureus, streptococcus pyognes, parasitter, mykotisk) (Weber S, 2024).
- Borreliose: Denne infeksjonen kan gi hudutslett og leddsmerter, men muskelsvakhet er mindre vanlig.
- Andre tilstander:
- Medikamentreaksjoner: Noen medisiner kan gi hudutslett og muskelsmerter som bivirkninger.
- Hypotyreose: Lavt stoffskifte kan gi muskelsvakhet og tretthet, men har vanligvis ikke hudutslett.
- Rhabdomyolyse (svært høy CK) utløst av:
- Genetisk myopati (RYR1-gen, MLIP-gen)
- Fettsyreoksydasjonsforstyrrelse (CPTII)
- Metabolske myopatier
Behandling av JDM
Behandlingen av JDM bør baseres på publiserte retningslinjer og veiledere (Enders FB, 2017; Papadopoulou C, 2023). Behandling er ofte krevende, men avgjørende for prognosen. Det er viktig å balansere behovet for tilstrekkelig immunsuppresjon med håndtering av bivirkninger, inkludert infeksjonsrisiko.
Medikamenter.
- Kortikosteroider. Initial behandling består ofte av høydose kortikosteroider, enten intravenøst eller peroralt (tabletter) (Orandi AB, 2021).
- Metotreksat. For å redusere behovet for kortikosteroider på sikt og forbedre effekten, kombineres behandlingen ofte med DMARDs, hvor metotreksat er førstevalget på grunn av gunstig bivirkningsprofil, mens ciclosporin eller mykofenolat er alternativer (Ruperto N, 2016).
- Rituksimab. Som andrelinje behandling ved utilstrekkelig effekt eller bivirkninger av metotreksat er biologiske legemidler, særlig rituksimab aktuelt (Sener S, 2023).
- IVIG. Også intravenøst immunglobulin (IVIG) brukes, spesielt mot hud-, muskel- og øsofagusmanifestasjoner (Lam CG, 2011).
- Andre. Cyklofosfamid (Sendoxan) brukes som tredjelinjebehandling ved alvorlig lungeaffeksjon. TNF-hemmere, abatacept, takrolimus, azathioprin, hydroksyklorokin (hudmanifestasjoner), tocilizumab og JAK-hemmere har vist effekt i noen rapporter og kan være aktuelle valg som utprøvende behandling i noen situasjoner (Papadopoulou C, 2023). CAR-T behandling har fremtidig potensiale til å bli aktuelt for de mest alvorlige tilfellene (Nicolai R, 2024).
Kalsinose har har vist seg å være behandlingsrefraktær i mange tilfeller, men rapporter tyder på en viss effekt av biologiske legemidler hos ca 50% (Sener S, 2023). Andre medikamentelle behandlingsmuligheter inkluderer IVIG, kortikosteroider, DMARDs, JAK-hemmere, bisfosfonater, diltiazem, natriumthiosulfat og lokalinjeksjon med kortikosteroider (Hoeltzel M, 2014). Annen behandling omfatter kirurgisk fjerning, knusing med sjokkbølger og bruk av karbondioksid-laser.
Lungemanifestasjon. Det er mangel på god dokumentasjon for optimal behandling i barnealder. I praksis foretrekkes nå kortikosteroider kombinert med rituksimab eller mykofenolat fremfor cyklofosfamid som har større generelt bivirkningspotensiale. I raskt progredierende og alvorlige tilfeller kan likevel cyklofosfamid foretrekkes, fordi medikamentvirkningen kommer relativt raskt (1-2 uker).
Tverrfaglig behandling. Fysioterapi er viktig for å tilpasse og følge opp et treningsprogram for å opprettholde muskelstyrke og funksjon. Psykologisk støtte bør også vurderes (Ardalan K, 2021).
Solbeskyttelse. For alle pasientene med hudsymptomer og/eller immunsuppressiv behandling anbefales solbeskyttelse.
Oppfølging
Oppfølging bør skje i samarbeid med et spesialisert senter. Medikamentdosene trappes gradvis ned, og seponering kan forsøkes dersom kortikosteroider er seponert og sykdommen har vært i remisjon i minst ett år. Under kraftig immunsuppresjon som under høye doser kortikosteroider, biologiske legemidler eller cyklofosfamid bør en alltid være oppmerksom på risikoen for infeksjon, for eksempel pneumocystis (PCP) i lunger.
Prognose
Høy sykdomsaktivitet kan føre til økt risiko for skader (Sanner H 2009). Selv etter 16 år har pasienter med JDM svakere muskler enn friske kontroller (Sanner H, 2010). I motsetning til dermatomyositt hos voksne er JDM ikke assosiert med malignitet.
Retningslinjer
Nasjonale retningslinjer for utredning, oppfølging og behandling
Britiske retningslinjer (Oldroyd AGS, 2022)
Kobayashi I, 2020 (Japan)
SHARE: Enders FB, 2016 (Utredning og behandling)
Litteratur
