ANDRE SYKDOMMER (REV 063-REV 077)
162 Histiocytose. Langerhans, Erdheim-Chesters, Multisentrisk retikulohistiocytose, Rosai-Dorfman (REV 063, REV 073)
Jan Tore Gran and Øyvind Palm
Kjennetegn på histiocytose
Vanskelige diagnoser å gjenkjenne.
De fleste typer begynner blant barn før 10 års alder, men voksen-former finnes også. Mange ulike manifestasjoner, fra harmløst til alvorlig.
Feber, vekttap, multiple skjeletthevelser, oftest i hodet/skallen, eksantem på hodet (Langerhans). Polyartritt i fingre (retikulohistiocytose), skjelettsmerter (Erdheim-Chester). Hepatosplenomegali (stor lever og milt), nodulære lungefortetninger (pulmonal histiocytose X). Lymfeknuteforstørrelser.
ICD-10: D76.3 (non-Langerhans), C96.0 Langerhans histiocytose (disseminert)
. CC BY-NC 3.0")
Definisjon
Histiocytose er sjeldne sykdommer der histiocytter (spesielle hvite blodlegemer: vevsmakrofager) ved en feil lagres i store mengder i forskjellige organer der de forårsaker skade. Eldre navn på sykdommene er histiocytose-X, Letter-Siwe sykdom, Hand-Schüller-Christian sykdom og diffus retikulo-endoteliose. Biopsier/histiologi kan vise “eosinofilt granulom”. Tidligere (WHO 1987) delte man sykdommen inn i tre hovedgrupper: Langerhans histiocytose (LCH), non-Langerhans og malign (ondartet) histiocytose. Senere har en gått over til en inndeling i fem hovedgrupper (se nedenfor) (Emile J-F, 2016).
Histiocytose håndteres vanligvis av hematolog. Revmatologens oppgave kan være differensialdiagnostiske vurderinger i utredningen.
Forekomst
Histiocytose er en sjelden sykdom. Den vanligste formen er Langerhans histiocytose med en insidens på 1-2/million per år (Tillotson CV, 2023).
Symptomer
Symptomer og undersøkelsesfunn varierer avhengig av hvilken type histiocytose som foreligger og hvilke organer som angripes hos den enkelte.
- Feber, tretthet og vekttap er generelle symptomene kan forekomme ved alle typer histiocytose.
- Smerte og hevelse i skjelettet er spesielt vanlig ved Langerhans celle histiocytose (LCH) og Erdheim-Chester sykdom.
- Retikulohistiocytose kan ligne revmatoid artritt (RA).
- Lymfadenopati: Kan ses ved LCH, Rosai-Dorfman sykdom (RDD) og hemofagocytisk lymfohistiocytose (HLH).
- Eksem eller noduli: Kan være et tegn på LCH eller andre former for histiocytose.
- Nekrobiotisk xantogranulom er en sjelden type non-Langerhans cellehistiocytose som ofte er assosiert monoklonal gammopati. Typisk er rødebrune eller gulaktige papler i huden og infiltrerte plakk. Området rundt øynene er det vanligst affiserte stedet. De fleste har en (IgG-type med kappa- og lambdakjeder) (Geoloaisa GL, 2021).
- Dyspne eller hoste: Kan indikere lungeinvolvering som er vanlig ved pulmonal Langerhans celle histiocytose.
- Diabetes insipidus: En tilstand med økt tørste og urinproduksjon på grunn av påvirkning av hypofysen. Forekommer ved LCH (Lin KD, 1998).
- Nevrologiske symptomer: Hodepine, svimmelhet eller balanseproblemer kan oppstå hvis histiocytose påvirker nervesystemet.
- Hud Vanligste sykdomsdebut er eksem, sår og subcutane noduli.
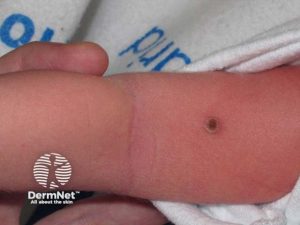
Diagnosen
Diagnosen stilles på bakgrunn av symptomer og undersøkelsesfunn. En benytter CT- MR- og PET/CT undersøkelser, men resultatet av vevsprøve (biopsi) er avgjørende.
Differensialdiagnoser
- Infeksjoner: Skjelettlesjonene kan ligne osteomyelitt. Hud-manifestasjoner kan se ut som eksem eller seboreisk dermatitt, og lungeaffeksjon kan forveksles med pneumoni eller tuberkulose. Det er viktig å vurdere bakterielle, sopp- og virusinfeksjoner (EBV, CMV og HIV).
- Kreft: LCH kan forveksles med kreftformer som leukemi, lymfom, Ewing sarkom og nevroblastom.
- Autoimmune sykdommer: Noen former for LCH med multiorganinvolvering kan etterligne autoimmune sykdommer som juvenil idiopatisk artritt eller systemisk lupus erythematosus.
- Andre benigne benlesjoner: Hvis LCH presenterer seg med isolerte skjelettlesjoner, er det viktig å skille det fra andre godartede skjelett-tumorer eller cyster.
- Lagringssykdommer: Enkelte lagringssykdommer kan føre til akkumulering av stoffer i vev, som noen ganger kan etterligne histiocyttiske infiltrater.
Behandling
Behandlingen av histiocytose varierer betydelig avhengig av den spesifikke typen, alvorlighetsgraden av sykdommen og involverte organer:
1. Langerhans celle histiocytose (LCH):
- Lokaliserte lesjoner: Kirurgisk eksisjon: For enkeltstående lesjoner i ben eller hud. Kortikosteroidinjeksjoner: Lokal behandling ved enkelte lesjoner. Stråleterapi kan vurderes ved utilgjengelige eller multiple benlesjoner.
- Multisystem sykdom: Kjemoterapi: Vinblastin, metotreksat, eller andre kjemoterapeutiske regimer. Kortikosteroider: For å dempe inflammasjon. Biologiske legemidler i noen tilfeller, spesielt ved refraktær sykdom. Supplerende behandling er avhengig av involverte organer (f.eks. hormonsubstitusjon ved hypofysesvikt) (PDQ Pediatric Treatment Editorial Board, 2025).
2. Andre histiocytose lidelser:
Behandlingen varierer og tilpasses den spesifikke typen histiocytose. Kortikosteroider, kjemoterapi, stråleterapi, og andre immunmodulerende medikamenter kan brukes.
Ulike typer histiocytose
. CC BY-NC 4.0")
1.) ”L” (Langerhans) gruppen
Langerhans celle-histiocytose (LCH)
Definisjon. Langerhans celle-histiocytose (LCH) er en sjelden tilstand ,med ukjent årsak, kjennetegnet ved unormale Langerhans (antigen presenterende) celler. Dette medfører en rekke symptomer fra hud, skjelett, lunger, lever, milt, lymfeknuter tarm eller hypofyse.
Forekomst: LCH rammer 1-2 per million og debuterer vanligvis i barnealder med 76 % av tilfellene under 10 år.
Symptomer. Symptomer og alvorlighetsgrad varier fra mild til alvorlig. Vanlige symptomer inkluderer utslett (ofte debutsymptom), feber, vekttap, multiple skjelettforandringer (ca. 80 %), oftest i hodet/skallen, men også i hofter/bekken, lårbein eller ribbein. Andre symptomer kan være eksem, sår, hepatosplenomegali, lymfadenopati og asymptomatiske, nodulære fortetninger i lungene (pulmonal histiocytose X).
Diagnosen bekreftes ved biopsi. Lesjoner farges med S-100 og CD1a. Elektronmikroskopi viser cytoplasmatiske Birbeck-granula.
Differensialdiagnoser er acrodermatitis enteropathica, acropustulosis hos nyfødte, kongenital candidiasis, eosinofil pustuløs follikulitt, incontinentia pigmenti, leukemi, lymfom, mastocytose, myelom og neonatal pustulær melanose,
Behandlingen varierer avhengig av alvorlighetsgrad og kan omfatte observasjon, medikamenter, kirurgi og strålebehandling.
Litteratur: Tillotson CV, 2024.
. CC BY-NC-ND 4.0")
Erdheim-Chester sykdom (ECD)
ICD-10: D76.3
Definisjon: Erdheim-Chester sykdom (EDS) er en sjelden, non-Langerhans histiocytose kjennetegnet av skjelettforandringer, ofte nært ankelleddene. Sykdommen er en multisystemsykdom som kan manifestere seg i flere organer. Illustrasjon: Leddnær skjelett-sklerose.
Historikk: ECD ble først beskrevet av Jakob Erdheim (1874-1937) og hans elev William Chester i 1930 som “lipoid granulomatose” (Chester W. Über Lipoidgranulomatose. Virchows Arch Pathol Anat 1930; 279: 561 – 6).
Etiologi: Sykdomsårsaken skyldes mutasjoner i BRAF-genet eller andre signal-molekyler som er assosiert med inflammatoriske cytokiner.
Epidemiologi: ECD er sjelden og den rammer menn hyppigere enn kvinner. Gjennomsnittlig debutalder er 55 år, men den kan oppstå i alle aldre (16-80år).
Symptomer:
- Smerter over tibia er et vanlig symptom, og biopsi vil vise fettrike histiocytter (CD68-positive). Sykdommen kan gi andre symptomer avhengig av hvilke organer som er involvert:
- Retroperitoneum: Fibrose
- Øyne: Eksoftalmus
- Lunger: Interstitiell lungesykdom (ILD)
- Hypofyse og hypotalamus: Hormonelle forstyrrelser
- Nyrer: “Hairy kidneys” (fibrose rundt nyrene)
- Hud: Xantelasmer
- Hjerte og aorta: Periaortal fortykkelse, kardiomyopati
Laboratorieprøver kan vise forhøyet CRP, SR og alkalisk fosfatase (ALP). Ved mistanke om hormonpåvirkning kan prolaktin LH, FSH, ACTH, GH eller TSH vise utsalg. Nyrefunksjonsprøver (kreatinin, GFR) gjøres for å vurdere nyrefunksjonen.
Bildediagnostikk:
- Skjelett: Røntgen og MR kan vise sklerose i de lange rørknoklene: typisk som symmetrisk osteosklerose.
- Bihuler: MR eller CT kan vise forandringer i sinus maxillaris.
- Nyrer og aorta: CT kan vise “hairy kidneys” og periaortal fortykkelse.
- Hjerte: Ekkokardiografi kan avdekke klaffesvikt, rytmeforstyrrelser, kardiomyopati (hyppigst i høyre ventrikkel) og periaortal fibrose med mulig innvekst i koronararteriene.
- PET/CT: Den beste metoden for oppfølging av sykdommen (Mazoor RD, 2013).
. CC BY-NC 4.0")
Ekkokardiografi kan vise insuffisiens i hjerteklaffer, rytmeforstyrrelser, kardiomyopati (hyppigst i høyre hjertedel) og tegn til fibrose periaortalt med innvekst og tranghet også i koronar-arterier.
Biopsi: Histologi viser skummakrofager eller xantogranulomatøs infiltrasjon i fibrinøst bindevev/stroma. Erdheim-Chester-celler uttrykker histiocyttmarkørene CD68 og CD163, men ikke CD1a eller S100 som skiller dem fra Langerhans-celler. Tester for BRAF og MAPK-ERK kan være relevante for behandlingsvalg.
Behandling: Behandlingen av ECD tilpasses alvorlighetsgraden. Asymptomatiske pasienter kan observeres uten medikamenter. Ved påvist BRAF V600E-mutasjon er vemurafinib (BRAF-hemmer) førstevalg. Interferon alfa er et alternativ. Ved andre mutasjoner (NRAS, KRAS, PIK3CA, MAP2K1 og ALK) kan cobimetinib vurderes. Kortikosteroider kan redusere inflammasjon, men påvirker neppe mortaliteten.
Litteratur: Jridi M, 2023; Haroche J, 2020; Mazoor RD, 2013; Midtvedt Ø, 2014
Systemisk Juvenilt xanthogranulom
Kjennetegn. Hudforandringer, CNS-affeksjon, lever og milt, øyet, svelg, muskler, Oftest barn under 1 års alder, sjeldnere hos eldre barn og voksne.
Litteratur: Hernandez-San Martin MJ, 2020; Collie JS, 2021.
. CC BY-NC-SA 3.0")
2.) “C” gruppen (Cutan (hud) og mucocutan (hud og slimhinner)
Multisentrisk Retikulohistiocytose (MRH)
Definisjon. Multisentrisk retikulohistiocytose (MRH) er en sjelden, systemisk inflammatorisk sykdom klassifisert som en non-Langerhans celle histiocytose klasse 2b (Zelger BWH, 1996).
Historikk. Sykdommen ble beskrevet som egen tilstand i 1937 av Weber og Freudenthal (Weber FP, 1937).
Epidemiologi. MRH er en sjelden sykdom som vanligvis rammer kvinner i alderen 50-60 år. Omkring 25 % har samtidig en malign sykdom.
Symptomer. MRH kjennetegnes av alvorlig artritt i hendene. Ofte ses hurtig innsettende, destruktiv og symmetrisk polyartritt. Sykdommen kan rammer distale interfalangealledd (DIP-ledd) og ledsages av karakteristiske hudlesjoner med et “perlekjede”-utseende rundt leddene. Xantomer ses hos 30 % av pasientene, og 30 % har lipidavvik i blodet. Andre symptomer kan inkludere myalgi og kontrakturer. Hos 60 % av pasientene begynner artritt et par år før andre symptomer.
Diagnose. Tidlig diagnostisering er essensielt for prognosen. En grundig klinisk undersøkelse og biopsi er viktig. Biopsi fra hud og synovium (leddhinne) kan vise multiple histiocytter med mulitnukleære kjempeceller og “matt-glass”-lignende eosinofilt cytoplasma. Histiocyttene inneholder PAS (periodic acid-Schiff) positivt materiale.
Behandlingen består av NSAIDS, kortikosteroider, metotreksat og biologiske legemidler (Tariq S, 2016).
")
Forløp og prognose. Sykdomsaktiviteten ved MRH kan variere og svinge over flere år. Tidlig behandling er viktig for å forhindre alvorlig, erosiv artritt som kan føre til leddskade og funksjonsnedsettelse.
Litteratur: Tariq S, 2016
Begrenset Retikulohistiocytose: Det finnes også en begrenset form for retikulohistiocytose.
Xantogranulom gruppen: Ulike former som kan angripe fra små barn til voksne. Enkelte eller mange røde-gule knuter i huden. 0,5-1,0 cm i diameter. Dersom også andre organer omfattes, innordnes sykdommen i “L” Gruppen (se ovenfor)
Hudaffeksjon med histiocytter i fettvev. Arvelig eller etter langvarig parenteral fettrik ernæring. Ofte relatert til myelodysplastisk syndrom (MDS) (Howard MR, 1993).
3.) “M” (Malign/kreft) Gruppen. Malign histiocytose
Sekundære former ved lymfom og leukemi. Subtype av akutt myeloid leukemi
Symptomer: Hoste, redusert appetitt, anemi, dyspne
Klinisk: Lunger, lymfeknuter, lever, milt og CNS kan affiseres ved histiocytt-infiltrasjon i biopsi.
Biopsi. Ulike primære former som ved vevsprøve skilles fra lymfom. Mer enn 20% blaster ved benmargsundersøkelse.

4.) “R” (Rosai-Dorfman) gruppe og forskjellige former som ikke angriper hud
Rosai-Dorfman sykdom, Sinus histiocytose med massiv lymfadenopati (SHML)
Definisjon: Sjelden sykdom med mange ulike manifestasjoner der blant annet lymfeknuter hovner opp. Biopsi er avgjørende for diagnosen og viser et typisk bilde med histiocytter som inneholder lymfocytter. Angriper vanligst barn og unge voksne.
Symptomer: Feber, nattesvette, tretthet, vekttap.
Medisinske undersøkelsesfunn: Varierende symptomer og forandringer: Hovne lymfeknuter, oftest på halsen, hudforandringer med fettatrofi, tetthet i nese og bihuler, trykk bak øyne og dobbeltsyn, forstørret milt. Osteonekrose
Diagnose: Symptomer og undersøkelsesfunn kan gi mistanke. Biopsi er avgjørende.
Differensialdiagnoser: GPA (Wegeners granulomatose). Pannikulitt. Langerhans histiocytose. Lymfom. Sarkoidose. Tuberkulose. IgG4 relatert sykdom
Behandling: Immundempende medikamenter. Kortikosteroider kan være tilstrekkelig hos noen.
Litteratur: Abla O, 2018
5.) “H” Gruppen. Hemofagocytisk lymfohistiocytose (HLH) og Makrofag aktiverings syndrom (MAS)
Cytokin storm med ukontrollert aktivering av lymfocytter og makrofager.
Se også makrofag aktiverings-syndrom (MAS)
Annet
Histiocytter ses også ved Kikutchi-Fujimoto sykdom (KFS) er en sjelden, benign, nekrotiserende lymfadenopati, oftest på halsen hos unge kvinner. Histologi (vevsundersøkelse ved biopsi) viser histiocytter og nekrotiserende non-maligne funn. Vennligst les om KFS i eget kapitel.
Litteratur
- Tillotson CV, 2023 (Langerhans)
- Emile J-F, 2016 (Klassifikasjon)
- Allen CE, 2015 (Langerhans: Behandling)
- Grischikowsky M, 2015 (Langerhans: Management)
- Tariq S, 2016 (multicentrisk retikulohistiocytose)
