ANDRE SYKDOMMER (REV 063-REV 077)
177 Loeys-Dietz syndrom (REV 063)
Øyvind Palm
- Karpaltunnelsyndrom, Bakers cyste, entesopati, lumbago, isjas, peritendinitt og kapsulitt i skulder er omtalt i egne kapitler.
ICD-10: Q87.4 (samme som Marfans syndrom)
Definisjon
Loeys-Dietz syndrom (LDS) er en sjelden, autosomal dominant arvelig sykdom i bindevev som disponerer for aneurismer i aorta og mindre arterier, selv i barnealder. Sykdommen kan ha likhetstrekk med Marfans syndrom, og regnes sammen med vaskulær form av Ehlers-Danlos syndrom og hereditært thorakalt aortaaneurisme (HTAD) (Isselbacher EM, 2016) som en av de arvelige ”Marfan-lignende” sykdommene. Loeys-Dietz syndrom er derfor ikke en autoimmun bindevevssykdom.
Revmatologens rolle er å skille LDS fra vaskulitt og ulike muskelskjelett sykdommer, noe som er av betydning fordi riktig oppfølging av Loeys-Dietz syndrom og operasjon om nødvendig kan forebygge alvorlige komplikasjoner og tidlig død. I Norge er Sunnås sykehus kompetansesenter.
Historikk
Sykdommen ble første gang beskrevet i 2005 (Loeys BL, 2005)
Forekomst
LDS defineres som en sjelden sykdom (færre enn ett tilfelle per 2.000 personer) (Det Norske Helse og Omsorgs-departementet). Spesifikke data for insidens og prevalens er ikke tilgjengelige, men Type 1 og type 2 (se nedenfor) er vanligst.
Sykdomsårsak
Ulike genmutasjoner forårsaker LDS og dens undergrupper:
- Type 1: Mutasjon i TGFBR1-genet
- Type 2: Mutasjon i TGFBR2-genet
- Type 3: Mutasjon i SMAD3-genet
- Type 4: Mutasjon i TGFB2-genet
- Type 5: Mutasjon i TGFB3-genet
Symptomer
Sykdommen kan manifestere seg på mange ulike måter, avhengig av hvilken subtype som foreligger. Symptomer kan omfatte (Gouda P, 2022):
- Kardiovaskulære: Aneurismer (ofte asymptomatiske), aortadisseksjon, hjertefeil
- Muskel- og skjelett: Skjelettabnormaliteter, hypermobile ledd, skoliose, kyfose
- Nevrologiske: Kraniosynostose (for tidlig lukking av skallebensømmene), utviklingsforsinkelse
- Gastrointestinale: Tarmdivertikler, tarmperforasjon
I noen tilfeller kan det også oppstå karakteristiske ansikts- og hudforandringer.
Utredning
Anamnese: Mange har hatt familiemedlemmer som brått døde i ung alder (Gouda P, 2022). Likevel påvises sykdommen oftest blant personer uten en familiehistorie.
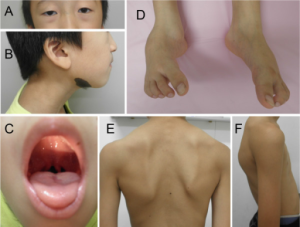
Klinisk. Det skjer en for tidlig sammenvoksing av skalleben i spedbarnsalder. Pasientene har ofte utvikler en karakteristisk ansiktsform med vid avstand mellom øyne. I tillegg ses ofte uvula bifida (spaltet drøvel) og ganespalte. I columna ses skoliose eller kyfose. Komplikasjoner med aneurismer i aorta og andre arterier er mest alvorlig. Thoraks med brystben kan utvikle deformiteter i oppveksten. Noen har en “Marfanoid habitus” med lange armer, ben, fingre og tær i forhold til kropp. Forekomsten og alvorlighetsgraden av aneurismer (med ruptur og blødningsrisiko) er individuelt forskjellig, men kan oppstå i barne-årene (Hyung-Tae S, 2015).
Laboratorium. Normale inflammasjonsparametere (utenom ved aneurismeblødning) og normale andre sykdomsmarkører.
Gentester: For å identifisere den spesifikke genmutasjonen; se genetikkportalen.no.
Bildediagnostikk: MR-angiografi, ultralyd Doppler/ekkokardiografi og CT for å undersøke blodårer og hjerte.
Diagnose
Diagnosen stilles basert på en kombinasjon av familiehistorie, symptomer, kliniske funn, bildediagnostikk og genetiske tester (genetikkportalen.no)
Differensialdiagnoser
Loeys-Dietz syndrom er sjelden og kan være vanskelig å diagnostisere. Det er viktig å skille LDS fra andre tilstander med lignende symptomer. Differensialdiagnoser er spesielt andre “Marfan-lignende” sykdommer:
- Marfans syndrom: arvelig bindevevssykdom som også kan gi utvidelse av aorta, lange lemmer og skoliose. Marfans syndrom kjennetegnes imidlertid ofte av høyere vekst, arachnodactyly (lange, slanke fingre og tær) og linseruksasjon i øyet, noe som er mindre vanlig ved LDS.
- Ehlers-Danlos syndrom: En gruppe arvelige bindevevssykdommer som kjennetegnes av hypermobile ledd, elastisk hud og økt blødningsrisiko. Noen typer EDS kan også gi utvidelse av aorta.
- Bicuspid aortaklaff: En medfødt hjertefeil der aortaklaffen har to i stedet for tre seil. Dette kan øke risikoen for utvidelse av aorta.
- Turner syndrom: En kromosomforstyrrelse som kun rammer kvinner og kan gi kortvoksthet, hjertemisdannelser og andre symptomer som kan overlappe med LDS.
- Homocystinuri: En sjelden, arvelig stoffskiftesykdom som kan gi symptomer som ligner på Marfans syndrom og LDS, inkludert lange lemmer og utvidelse av aorta.
- Noonan syndrom: En genetisk tilstand som kan gi hjertemisdannelser, kortvoksthet og andre symptomer som kan ligne på LDS.
- HTAD (Heritable thoracic aortic disease). Genetiske thorakale aortaaneurismer forårsaket av mutasjoner som også kan medføre aortadilatasjon og aneurismer.
Komplikasjoner
De mest alvorlige komplikasjonene inkluderer aortaaneurismer, oftest i ascendens og i aortabuen, samt aortadisseksjon (Gouda P, 2022).
Behandling
Ingen helbredende behandling for LDS er tilgjengelig.
- Kirurgi: Operasjon kan være nødvendig for å reparere eller erstatte utvidede blodårer. Dersom ett eller flere aneurismer øker i størrelse, kan operasjon for å forebygge ruptur bli nødvendig.
- Blodtrykkskontroll: Streng blodtrykkskontroll er viktig for å redusere belastningen på blodårene.
- Begrensning av fysisk aktivitet: Intensiv trening bør unngås, men vanlig fysisk aktivitet anbefales.
- Oppfølging: Regelmessige kontroller av blodårene er viktig for å oppdage aneurismer tidlig. Hvis en arterie utvider seg, kan målinger minst hvert halvår være aktuelt, ellers årlig i en lengre periode.
Litteratur
- Gouda P, 2022
- Yarate YA, 2015 (Aortadilatasjon, utvidelse av hovedpulsåren) blant barn og unge
- MacCarric G, 2014 (Dagose og management)
- Loeys BL, 2005 (original beskrivelse av syndromet)
- Kompetansesenter: Sunnås Sykehus
