ANDRE SYKDOMMER (REV 063-REV 077)
195 Paget Sykdom (Osteitis deformans) (REV 073)
Øyvind Palm and Jan Tore Gran
Kjennetegn på Pagets sykdom i skjelettet
Varierende skjelettsmerter og påvisbar fortykket bensubstans i bekken, femur, lumbal-columna, hodeskalle og/eller tibia.
Økt frakturrisiko
Genetiske former forekommer (ofte fra England, Skottland, Sentral-Europa eller Hellas).
ALP (alkalisk fosfatase) og P1NP (benmetabolisme) er økt i blodet.
ICD-10: M88.9

Definisjon
Paget sykdom i skjelettet er en non-inflammatorisk, metabolsk skjelettsykdom som medfører unormalt økt omsetting av ben, med høy aktivitet av både osteoklaster (celler som bryter ned benvev) og osteoblaster (celler som bygger opp benvev). Det nydannede benvevet er midlertid ikke normalt, men fortykket, deformert og svekket. Mange pasienter med Paget sykdom blir vurdert av revmatolog, men oppfølgingen er ofte hos endokrinolog eller i et samarbeid.
Sykdommen har ulike symptomer. Omtrent 3/4 er symptomfrie i lang tid, mens skjelettsmerter ellers er vanlig (Rianon NJ, 2020). Paget sykdom i skjelettet som er omtalt her må ikke forveksles med Pagets sykdom i brystvorten.

Historie
Sykdommen (Paget sykdom i skjelettet) ble opprinnelig beskrevet som “Chronic Inflammation of the Bones; osteitis deformans” av Sir James Paget (1814-1899). Hans navn er også knyttet til andre medisinske tilstander som intraductal brystkreft (Pagets disease of the nipple), Paget–Schroetter sykdom (dyp venetrombose i arm) og Pagets abscess (abscess-residiv).
Den initiale teorien om at sykdommen var en inflammasjon ble etter hvert forlatt. På 1920-tallet og 1930-tallet viste patologiske studier at sykdommen var en forstyrrelse i benvevets remodelleringsprosess.
På slutten av 1900-tallet og tidlig 2000-tallet klarte forskere å identifiserte mutasjoner i SQSTM1-genet som en viktig genetisk årsak.
Epidemiologi
Pagets sykdom i skjelettet rammer menn litt hyppigere enn kvinner (1,5:1) og debuterer vanligvis etter 50-årsalderen. Insidensen omtrent dobles for hver senere dekade. Paget sykdom er mest vanlig i England, Skottland, Sentral-Europa og Hellas (prevalens etter 50 års alder på hele 2-9%), men sjelden i Skandinavia og i Asia. Data indikerer at sykdommen er blitt sjeldnere over tid (Cook MJ, 2021; Corral-Gudino L, 2013).
Patogenese
Pagets sykdom i skjelettet er preget av lokalisert økt benmetabolisme. Først skjer det en osteoklast-mediert nedbrytning av beinvev, etterfulgt av en kompensatorisk osteoblastisk nydannelse av ben. Denne prosessen fører til at skjelettet blir ustrukturert og skjørt, med økt risiko for brudd. Noen individer har en genetisk disposisjon for Pagets sykdom (Choi YJ, 2022).
Lokaliseringen av sykdommen kan delvis være relatert til somatiske områder med fysisk belastning (f eks “Billiard-player’s fingres”) (Solomon LR, 1979).
Utenom genetisk disposisjon, antas miljøfaktorer å være av betydning for sykdomsutviklingen. Virus (spesielt paramyxovirus som meslingvirus) og vitamin-D-mangel er undersøkt i flere studier (Barker DJ, 1974), men med motstridende resultater (Matthews BG, 2008).
Genetikk
Familiære former for Pagets sykdom forekommer hos 5-40% av pasientene. Pagets hos førstegradslekting øker risikoen for sykdommen 7-10 ganger (Lugi G, 2022). Disse har ofte tidligere sykdomsdebut enn de med sporadiske tilfeller.
SQSTM1 (Sequestosome 1) er det mest studerte og hyppigst muterte genet ved Pagets sykdom. Mutasjoner i SQSTM1-genet finnes hos 20-40 % av pasienter med familiær Pagets sykdom og hos rundt 5-10 % med sporadisk (ikke-arvelig) form. SQSTM1-genet koder for et protein som regulerer funksjonen til osteoklaster. Mutasjoner fører til at osteoklastene blir overaktive (Lugi G, 2022).

Symptomer
Omtrent 75% av pasientene med Pagets sykdom er lenge asymptomatiske, og diagnosen stilles ofte tilfeldig ved røntgenundersøkelse eller forhøyet alkalisk fosfatase (ALP) i blodet. Sykdommen kan ramme ett eller flere områder av skjelettet (Bouchette P, 2023 sp). Blant symptomene er skjelett-smerte det vanligste.
Skjelettsymptomer:
- Ømhet eller smerte dypt i skjelettet, ofte verre om natten.
- Deformiteter i skjelettet
- Forstørret og fortykket skalle (“hatten blir for trang”) og kjeve
- Økt krumning i tibia
- Kyfose og skoliose
- Økt varme over berørte områder på grunn av økt blodtilførsel.
- Frakturer på grunn av svekket beinstruktur.
- Artrose når når sykdommen påvirker leddnære områder.
Andre symptomer:
- Hørselstap, vertigo og tinnitus ved påvirkning av skjelettet rundt det indre øret (vestibularis-nerven: entrapment).
- Nevrologiske komplikasjoner: Perifer nevropati, isjas og inkontinens for urin og avføring ved cauda equina syndrom (klemming av nerver i nedre del av ryggmargen). Migrene.
- Hjertesvikt (sjelden): På grunn av økt belastning på hjertet ved økt blodtilførsel til skjelettet.
Undersøkelser
Anamnesen omfatter genetisk disposisjon og aktuelle symptomer.
Klinisk gjøres en grundig vurdering av skjelettet, inkludert hodeskallen.
Laboratorieprøver (patologi)
- Forhøyet alkalisk fosfatase > 20% over øvre referanseområdet (helst benspesifikk ALP) og urinsyre (økt cellemetabolisme) . Differensialdiagnostisk: Sjekk også vitamin D og PTH (utelukke sekundær hyperparatyreoidose).
- Økte markører for benmetabolisme (P1NP ; procollagen type 1N-terminal propeptide).
- Normale inflammasjonsparametere, serumkalsium og -fosfat.
- Sekundær hyperparathyreoidisme ses hos 10% av pasientene (kalsiummangel ved høy skjelettmetabolisme).
- Urin: Økt nivå av hydroksyprolin, C-telopeptid og N-telopeptid som er markører for nedbrytning av bensubstans.
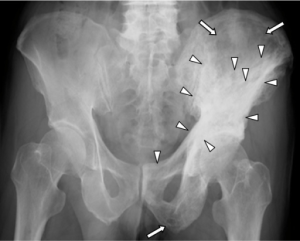
Bildediagnostikk.
De radiologiske forandringene ses oftest i bekkenet (70%), femur (55%), lumbalcolumna (53%) hodeskalle (42% og tibia (32%) (Ralston SH, 2012).
Røntgen og CT: Kan vise typiske forandringer som fortykket og deformert beinvev (osteolyse i tidlige stadier, osteosklerose senere), frakturer og artrose.
PET/CT: Kan vise økt opptak av 18FDG i skjelettet som kan ligne på malignitet.
Biopsi: Benbiopsi er vanligvis ikke nødvendig.
Genetisk testing: Man kan påvise disposisjon for sykdommen (Makaram NS, 2021).
Differensialdiagnoser
- Erdheim-Chester sykdom (en histiocytose): En sjelden tilstand som kan gi skjelettforandringer, men typisk med symmetrisk affeksjon av lange rørknokler, i motsetning til Pagets sykdom.
- Hypervitaminose D: Overdosering av vitamin D kan føre til økt benomsetning og hyperkalsemi som i sjeldne tilfeller kan gi radiologiske funn som ligner på Pagets sykdom.
- Hypoparathyroidisme: Lavt parathormon som kan gi økt bentetthet, men som oftest gir et annet radiologisk bilde enn Pagets sykdom.
- Metastaser: Spredning av kreft til skjelettet kan gi både osteolytiske (myelomatose, brystkreft, nyre- og thyreoidea- cancer) og osteoblastiske/sklerotiske (prostatacancer, brystkreft) som i noen tilfeller kan ligne på Pagets sykdom.
- Osteosklerose / osteopetrose av andre årsaker:
- Albers-Schönberg sykdom, Pycnodysostosis, Van Buchem sykdom (kjeve og ekstremiteter) og Sclerosteosis er sjeldne genetiske sykdommer
- Myelofibrose: arrvev erstatter den normale benmargen
- Fluorose: Overdreven inntak av fluor
- Renal osteodystrofi: Hos personer med kronisk nyresykdom
- Mastocytose: Økt mastcelleaktivitet som i noen tilfeller kan gi skjelettforandringer, men oftest med et annet klinisk bilde enn Pagets sykdom.
- Osteomyelitt (osteoskleroserende form): Fenne formen for osteomyelitt til områder med økt bentetthet (sklerose) på røntgenbilder. Dette kan få det til å ligne på det fortykkede, tettere beinet som sees ved Pagets sykdom.
- Osteosarkom: Noen undergrupper kan forårsake områder med økt bentetthet eller en blanding av økt og redusert tetthet som kan overlappe med utseendet til Pagets sykdom.
- Sarkoidose: Kan involvere skjelettet og gi lesjoner som ligner på Pagets sykdom.
- Tuberøs sklerose: En genetisk sykdom som kan gi hamartomer i ulike organer, inkludert skjelettet, som i sjeldne tilfeller kan ligne på Pagets sykdom.
Behandling
Behandlingen av Pagets sykdom i skjelettet skal primært redusere smerte, bremse sykdomsprogresjonen og minimere komplikasjoner. Behandling er ofte ikke nødvendig hos asymptomatiske pasienter eller ved normal ALP i blodprøver. Behandlingsindikasjoner inkluderer:
- Skjelettdefekter, særlig i vektbærende strukturer
- Skalle-deformiteter
- Raskt progredierende skjelettforandringer
- Sykdomsrelatert smerte
Medikamenter:
- Smertebehandling med paracetamol, NSAIDs eller medikasjon mot nevropati kan være aktuelt.
- Bisfosfonater (som også brukes i osteoporose-behandling). Disse legemidlene er førstevalget i medikamentell behandling (Singer FR, 2020). De hemmer osteoklastenes aktivitet og dermed nedbrytningen av benvev. Vanlige bisfosfonater inkluderer zoledronsyre (Aclasta 5 mg iv), pamidronat, risendronat og alendronat. Zoledronsyre gis ofte intravenøst, og har vist seg svært effektivt. Den medikamentelle behandlingen av sykdommen monitoreres med måling av alkalisk fosfatase i serum.
- Kalsitonin eller denosumab (antiresorptiv behandling): Alternativer til bisfosfonater, men mindre brukt på grunn av lavere effektivitet. Kan brukes hos pasienter som ikke tolererer bisfosfonater (Ralston SH, 2019).
Ortopedisk kirurgi:
- Kan være nødvendig for å korrigere deformiteter, behandle brudd eller utføre leddprotesekirurgi.
- Spesielt aktuelt ved komplikasjoner som frakturer eller artrose.
Annet: Kalsium og D-vitamin kan ha symptomatisk virkning.
Prognose
Ubehandlet kan Pagets sykdom føre til uttalte skjelettdeformiteter over tid. Sykdomsforløpet kan deles inn i fire faser.
- Initialt øker osteoklastaktiviteten.
- Hybrid osteoklast/osteoblast-prosess.
- Osteoblastaktivitet
- Malign degenerasjon (Rianon NJ, 2020).
Det er en liten risiko (mellom en av 500 og en av 1000 tilfeller) for utvikling av skjelettkreft, som osteosarkom og kjempecelletumorer.
Retningslinjer
Litteratur
