ANDRE SYKDOMMER (REV 063-REV 077)
218 Sneddon syndrom (REV 038, REV 103)
Øyvind Palm
Kjennetegn på Sneddons syndrom
Cerebral iskemi og slag eller retinal arterieokklusjon hos kvinner i 20-40-årene.
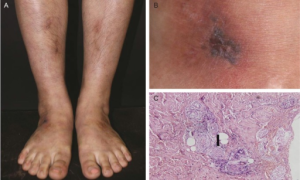
Livedo racemosa (marmorert hud med obstruksjon av kar) med typiske histologiske forandringer.
Antifosfolipid syndrom relaterte tilfeller eller familiære tilfeller med genmutasjoner.
- Differensialdiagnoser ved vaskulitt, kutan vaskulitt, hjerneslag og tromboembolier er beskrevet i et egne kapitler.
ICD-10: M30.8
Definisjon
Sneddons syndrom er en non-inflammatorisk trombotisk vaskulopati som påvirker små- og mellom-store arterier. Sykdommen fører til kroniske hudmanifestasjoner og risiko for multiple hjerneslag i ung alder (Wu S, 2014). Den rammer oftest voksne kvinner i alderen 20-40 år. Ulike sykdomsårsaker har vært foreslått uten at en klar konsensus er oppnådd (Starmann NLP, 2021).
Tilstanden er ikke en autoimmun revmatisk vaskulitt. Syndromet er tidligere inndelt i ulike former:
- Assosiert med antifosfolipidantistoff eller systemisk lupus (SLE).
- Trombotisk form (Schellong SM, 1997).
- En idiopatisk generalisert type (ukjent årsak).
Blant disse dekkes type 1 av antifosfolipidantistoff syndromet (primært eller sekundært). Den generaliserte, idiopatiske typen er mer særegen, og det er påvist genetiske assosiasjoner (Greisenegger AK, 2021). Revmatologens oppgave ved Sneddons syndrom er å skille tilstanden fra inflammatorisk revmatisk sykdom og bidra til diagnostisering.
Historie
Sammenhengen mellom hudsymptomene livedo racemosa og de cerebrovaskulære manifestasjonene ble først beskrevet av Kimming in 1959 (Kimming J: Arteriolopathie: livedo rasemosa. Dermatol Wochenschr. 1959, 139: 211). Sneddon rapporterte seks tilfeller i 1965 (Sneddon IB, 1965).
Forekomst
Den estimerte insidensen av Sneddon syndrom er fire per million årlig. Sykdommen er hyppigst hos kvinner i alderen 20-40 år. Blant hospitaliserte pasienter med hjerneslag er forekomsten beregnet til 0,25-0,50 % (Wu S, 2014).
Sykdomsårsak
Det finnes flere undergrupper av Sneddons syndrom som trolig har ulik patogenese. Genmutasjoner er påvist i noen familiære tilfeller. Disse mutasjonene kan forårsake et lignende sykdomsbilde (med slag og hudforandringer) som DADA2, juvenil PAN blant barn, eller CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) hos middelaldrende.
- Genmutasjon i adenosin deaminase 2 (ADA2) genet/cat eye syndrome chromosome region- candidate-1-genet (CECR1) ble påvist i en angrepet portugisisk familie i 2014. Deres symptomer var slag i tidlig voksen alder, ulcerasjoner på underekstremiteter og intermitterende feber. Denne formen er senere påvist i flere familier og overlapper da med “DADA2/juvenil PAN“.
- Homozygot NOTCH3-mutasjon: Mutasjoner i NOTCH3 er årsaken til CADASIL som er en cerebral småkarsykdom med progressive iskemiske manifestasjoner fra 40-50 års alderen (Greisenegger AK, 2021).
- Antifosfolipid-antistoff som medfører økt tromboemboli-risiko og til dels er relatert til systemisk lupus (SLE) forekommer hos 30-40%.
- Idiopatiske (uforklarte) former er blitt sjeldnere etter at genanalyser ble tilgjengelige.
Symptomer
- Hudforandringer (livedo retikularis/racemosa, ulcerasjoner) kan forutgå de nevrologiske symptomene med 10 år. De begynner ofte på armer og ben og forverres i kulde.
- Nevrologiske manifestasjoner omfatter hodepine (det vanligste symptomet allerede fra barne- og ungdomsalderen), migrene, hukommelses-svikt (amnesi) og andre kognitive besvær, talevansker (afasi), TIA og hjerneslag.
- Organskader på grunn av iskemi kan pasientene også utvikle skader i hjerte, nyrer og øyne.
- Andre nevrologiske symptomer kan inkludere transient amnesi, afasi, pareser og migrene/hodepine, TIA og hjerneslag. Hypertensjon kan debutere hos barn, men oftere i alderen 20-40 år, og rammer oftest kvinner.
- Utbredt livedo racemosa er ofte første symptom allerede i barne-ungdomsalderen. Ikke all litteratur skiller mellom livedo retikularis og racemosa. Til forskjell fra livedo reticularis (reversible kar-spasme) er livedo racemosa forårsaket av okklusjoner i små og mellomstore kar.
Undersøkelsesfunn
Anamnese omfatter spørsmål om familietilfeller med tidlig slag og hudforandringer, samt symptomer på assosiert systemisk lupus (SLE).
Klinisk undersøkelse: Dokumenterer hudsymptomene. Uttalt livedo med ulcerasjoner kan forekomme, særlig på underekstremitetene. Det er også økt forekomst av hypertensjon og patologiske hjerteklaffer. Undersøkelsen bør inkludere vurdering av tegn på tromboembolier i aktuelle organer og tegn på systemisk lupus.
Hudbiopsi: På grensen mellom dermis og subcutis kan man påvise tegn til inflammasjon i endotel, subendotelial celleproliferasjon og fibrose i okkluderte arterier.
Blodprøver omfatter antifosfolipid antistoff (lupus antikoagulant, anti-kardiolipin, anti-beta-2GP) som påvises hos ca. 50 % av pasientene. Suppler med ANA og ANCA (differensialdiagnostisk vaskulitt i små- og mellomstore kar).
Bildediagnostikk utføres med MR-undersøkelse av hjernen ved tegn til cerebrale manifestasjoner som leukoencefalopati, infarkter og aneurismer. Tegn til dyp venetrombose kan vurderes med ultralyd. Ekkokardiografi kan påvise patologiske hjerteklaffer.
Gentest for mutasjon av adenosin deaminase 2 (ADA2)/CECR1 og NOTCH3 er aktuelt (Genetikkportalen.no). Klinisk genetisk oppfølging er nødvendig hvis det gjøres funn.
Nevrolog bør konsulteres for kartlegging av eventuelle nevrogene symptomer og utfall
Øyelege bør konsulteres ved synsforstyrrelser (sentral retinal arterie- eller vene okklusjon, retinal neovaskularisering, homonymt synsfeltutfall og internukleær oftalmoloplegi).
Differensialdiagnoser
Her er noen av de viktigste differensialdiagnosene og hvorfor de kan ligne på Sneddons syndrom:
- Antifosfolipid syndrom (APLS): Kan også gi livedo retikularis og økt risiko for blodpropp, men er ofte assosiert med andre symptomer som gjentatte spontanaborter og trombocytopeni.
- Cerebral iskemi av andre årsaker: For eksempel aterosklerose, hjerteemboli og vaskulitt.
- CNS-vaskulitt (PACNS): Inflammasjon i blodårene i hjernen og kan gi livedo retikularis og neurologiske symptomer, men har ofte et mer akutt forløp og andre funn ved nevrologisk undersøkelse.
- Cutis marmorata: Dette er en normal respons på kulde som gir et marmorert utseende på huden, men forsvinner når man blir varm. I motsetning til livedo retikularis ved Sneddons syndrom er det forbigående og ikke assosiert med andre symptomer.
- Kryoglobulinemi: kjennetegnes av tilstedeværelse av kryoglobuliner (proteiner som klumper seg sammen i kulde) i blodet og kan gi livedo retikularis og andre symptomer som leddbetennelse og nyreproblemer.
- Polyarteritis nodosa (PAN): systemisk vaskulitt som kan gi livedo retikularis, neurologiske symptomer og andre symptomer som feber, vekttap og magesmerter.
Behandling
Behandlingen av Sneddons syndrom er primært symptomatisk og rettet mot å forebygge tromboemboliske hendelser, samt håndtere assosierte symptomer. Det finnes ingen kurativ behandling.
Antikoagulasjon anbefales som initial behandling og forebyggende mot tromboembolier. I tilfeller med antifosfolipidantistoffer har direktevirkende antikoagulantia (DOAK) usikker effekt, slik at warfarin (Marevan) er mest aktuelt. Doseringen bør være relativt høy, slik at INR er >3. Effekten er da bedre enn acetylsalisylsyre (ASA) eller lavere dosert Marevan (Francès C, 1999).
Acetylsalisylsyre (ASA): Antiplatelet midler, som acetylsalisylsyre (ASA) eller klopidogrel, kan også vurderes, særlig i kombinasjon med antikoagulasjon eller som et alternativ ved kontraindikasjoner.
Vasodilatorer, som kalsiumkanalblokkere (f.eks. nifedipin), kan være nyttige for å forbedre hudens mikrosirkulasjon og lindre symptomer som livedo reticularis, men forebygger ikke cerebrale manifestasjoner.
Immunsuppressive legemidler som kortikosteroider og DMARDs brukes vanligvis ikke (Akbal A, 2010).
Oppfølging: Regelmessig overvåking av antikoagulasjonsnivåer og nevrologisk status er essensielt.
Prognose
Kronisk cerebral iskemi og sekvele etter tromboembolier kan bidra til tidlig demens. En mortalitet på 9,5% er rapportert etter 6,2 års observasjon i eldre studie (Zelger B, 1993).
Litteratur
