ANDRE SYKDOMMER (REV 063-REV 077)
134 Amyloidose, amyloid avleiringssykdom (REV 073)
Amyloidose
Øyvind Palm
Kjennetegn på amyloidose
Uspesifikke symptomer som polynevropati, kardial svikt, nyresvikt, “drop-head”.
Glatt, fortykket og lett-blødende hud.
Makroglossi, hepatomegali.
Proteinuri.
Biopsi er diagnostisk (spesialfarging).
AL amyloidose ble tidligere kalt primær type, AA amyloidose sekundær type.
Diagnosekoder ICD10: E85.8
Definisjon
Systemisk amyloidose er en gruppe sykdommer karakterisert ved ekstracellulær avleiring av amyloid, et uoppløselig stoff med en fibrillær struktur. Amyloid akkumuleres i vev og organer, da det ikke kan brytes ned enzymatisk. Nesten alle vev og organer kan affisere. Amyloidose kan klassifiseres i flere undergrupper:
- AA-Amyloidose (sekundær amyloidose) kan oppstå ved kronisk inflammasjon ved revmatiske sykdommer. AA-amyloidose er dermed relatert til langvarig systemisk inflammasjon ved sykdommer som familiær middelhavsfeber, juvenil artritt, revmatoid artritt, Bekhterevs, Muckle Wells syndrom og inflammatorisk tarmsykdom.
- AL-amyloidose er en primær form (systemisk lettkjede-amyloidose), som kan ha sykdomstrekk som kan forveksles med revmatisk sykdom (vennligst se differensialdiagnoser nedenfor).
- Lokalisert amyloidose, f eks. makulær amyloidose defineres ved at amyloid produseres og avleires i et isolert område av kroppen. Det finnes ikke monoklonale lette kjeder i blodet, og prognosen er god.
- Andre typer: vennligst se tabellen nedenfor.
Utredning og oppfølging av amyloidose gjøres av spesialist innen det/de mest berørte organene, eventuelt også hos hematolog. Revmatologen bør kunne gjenkjenne symptomene og skille dem fra revmatiske bindevevssykdommer, vaskulitt og artritt.
Historie
Rudolf L. K. Virchow (1821-1902, Berlin og Würzburg). Den første som beskrev leukocytose, leukemi og embolisme utgått fra trombose. Han betegnet i 1854 strukturer som amyloid (“som ligner stivelse”). Tidligere tilfeller av amyloidose er likevel antakelig beskrevet (Kyle RA, 2001).
 (2012). CC BY-NC 3.0")
Patogenese
Ved amyloidose syntetiseres unormale ekstracellulære, misfoldede proteiner som aggregerer og avleires i ulike organer. AL amyloidose stammer vanligvis fra en plasmacellesykdom hvor en liten klon i benmargen langsomt og progressivt utskiller ustabile lette kjeder av immunglobuliner (Al Hamed R, 2021). M-komponenten er vanligvis lav, men amyloidose kan også forekomme ved myelomatose. Ved AA-amyloidose er amyloid protein A (et akuttfaseprotein) en forløper (prekursorprotein) som kan påvises i serum i form av SAA (Chiti F, 2017; Husby G, 1992).
Epidemiologi
En svensk studie fant en årlig insidens på 1-2 per 100 000, med AL-amyloidose som den vanligste typen. Forekomsten er høyest blant 60-80-åringer (Hemminki K. 2012). Fordeling av amyloidose har endret seg de siste tiårene, med en reduksjon i AL-amyloidose og en relativ økning i AA-amyloidose (de Asua D, 2014). Familiær transthyretin-assosiert amyloidose (ATTR) er en sjeldnere type som utgjør 10- 20% (Bustamante JG, 2021).
Klassifikasjon
Amyloidose klassifiseres etter amyloidproteinet som misfoldes i patogenesen, sykdomsforløpet og den kliniske presentasjonen. Det finnes nær 40 ulike typer, men hovedgruppene er primær AL-amyloidose, sekundær amyloidose (AA), familiær amyloidose og ß2-mikroglobulin-relatert amyloidose. Noen vanlige typer er vist i tabellen nedenfor (modifisert etter de Asua D, 2014). Dessuten er det ved inklusjonslegeme myositt funnet amyloid-avleiringer av Alzheimer-type (A13) i inklusjonslegemer og vakuoler.
| Betegnelse | Amyloid-protein | Klinisk syndrom |
| AL (primær) | Monoklonale lette immunglobulin-kjeder | Monoklonal gammopati: myelomatose (kappa eller lambda) |
| AA (sekundær): RA, JIA, Bekhterevs, familiær middelhavsfeber, Behcets | Serum amyloid A protein (Apo-SAA) | Inflammasjons-assosiert |
| Cerebral amyloid | β-protein | Alzheimer demens, trisomi-21 (Downs syndrom), iatrogen Creutzfeldt Jakobs sykdom |
| Familiær amyloidose | Mutant transthyretin, A1-apoprotein, gelsolin, fibrinogen, lysozym med flere | Familiær polynevropati, kardiomyopati eller nefropati |
| Transthyretin-relatert amyloidose | Vill-type transthyretin | Senil restriktiv kardiomyopati |
| Islet amyloid polypeptide (IAPP) | Islet amyloid peptid | Diabetes type II |
| Hereditær | beta2- mikroglobulin | Kronisk dialyse |
Symptomer
Amyloidose kan gi en rekke ulike symptomer, avhengig av hvilke organer som er rammet. Sykdommen kan derfor være vanskelig å diagnostisere i tidlig fase, da symptomene ofte er uspesifikke og kan forveksles med andre tilstander. Dette kan føre til forsinket diagnose og behandling (Vaxman I, 2020).
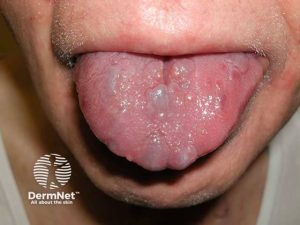
Vanlige symptomer omfatter blant annet kardial svikt med bevart ejeksjonsfraksjon (HFpEF) og proteinuri (nyremanifestasjon). I tidlig fase kan hjerteproblemer som angina pectoris, ortostatisk hypotensjon og hjerterytmeforstyrrelser (arytmier) forekomme (Deshai HV, 2010). Amyloidavleiring i organer kan føre til hepatomegali, makroglossi og redusert funksjon i spyttkjertler (sicca-symptomer). Perifer nevropati, vekttap (på grunn av tarmaffeksjon) skyldes amyloidavleiringer. Hevelse over skulderledd (amyloide skulderputer) skyldes også amyloid-avleiringer (Liepnieks JJ, 2008). Utmattelse er også vanlig (Al Hamed R, 2021).
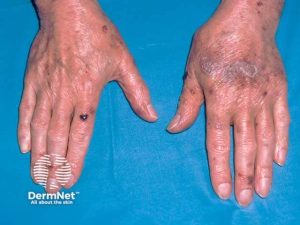
Andre symptomer kan være voksaktig fortykkede fingre og ekkymoser under en lett-blødende hud, spesielt rundt øynene. Hjernen affiseres vanligvis ikke av amyloidose.
Tabell over symptomer ved amyloidose, avhengig av organaffeksjon: (Ihne S, 2020):
| Hjerte/kardialt | Dyspne, perifere ødemer, anasarka, pleuravæske, perikardvæske , palpitasjoner, rytmeforstyrrelser synkoper, hypotensjon eller regress av hypertensjon, bradykardi |
| Nyrer | Ødem, skummende urin / proteinuri (eventuelt nefrotisk syndrom), nyresvikt |
| Lever | Hepatomegali, forhøyet alkalisk fosfatase, ascites |
| Mage-tarm | Dysfagi, redusert appetitt, vekttap, kvalme, postprandial metthetsfølelse, meteorisme, diare, obstipasjon, gastrointestinal bløding |
| Nevrologisk; perifere og autonome nerver | Polynevropati med progressiv, symmetrisk, affeksjon av aksonale / små fibre, varierende manifestasjoner. Vegetativ dysregulering (ortostatisk hypotensjon). Intestinal motilitets forstyrrelser, urin retensjon, erektil dysfunksjon |
| Øye | Tørre øyne, uklart syn, glaukom, retinal angiopati |
| Hud, bløtdeler og andre manifestasjoner | Makroglossi, heshet, koagulasjonsforstyrrelser, purpura/kutan hemoragi (ofte periorbitalt), karpal tunnel syndrome, hovne ledd (ikke artritt), splenomegali, myasteni, fatigue, biceps sene ruptur, lumbal spinal stenose. “Dropped-head syndrome ” (svakhet i nakke-ekstensorer) rapportert som debutsymptom (Differensialdiagnose: myositt). |
Diagnose
Hos pasienter med AL-amyloidose har 69% allerede manifestasjoner i flere organer ved diagnosetidspunktet. Tidlig diagnose er derfor avgjørende for å kunne behandle den underliggende plasmacelledyskrasien og som for AA-amyloidose, forhindre progressiv organskade (Obici L, 2005; Al Hamed R, 2021).
. CC BY 2.0")
Amyloidoses varierte og ofte uspesifikke symptomer gjør det utfordrende å diagnostisere tilstanden tidlig. Klinisk mistanke vekkes gjerne på bakgrunn av symptomer og påvisning av proteinuri. For å bekrefte diagnosen er det nødvendig med biopsi og histologisk undersøkelse.
Blodprøver kan gi viktig informasjon om organfunksjon og forekomst av proteiner assosiert med amyloidose. Følgende prøver kan være aktuelle: celletellinger, lever – og nyrefunksjonsprøver, albumin, samt NTPro-BNP og troponin T. Proteinelektroforese.
I tillegg kan laser capture microdissection og tandem massespektrometri (LCM-MS) brukes for subtyping av amyloid, en metode som er mer spesifikk enn antistoffbaserte metoder og immunhistokjemi (Wisniowski B, 2020).
Urin: Urinprøver undersøkes for proteinutskillelse og protein/kreatinin-ratio. Urinelektroforese kan også være nyttig.
EKG kan avdekke rytmeforstyrrelser hos pasienter med kardiale manifestasjoner av amyloidose.
Biopsi er avgjørende for å stille en sikker diagnose. Vevsprøver kan tas fra ulike steder, og prosedyren er vanligvis ukomplisert. Det er imidlertid viktig å være oppmerksom på at pasienter med amyloidose kan ha økt blødningsrisiko. Vanlige biopsisteder er abdominalt fettvev (sensitivitet >80%), rektum-slimhinne eller små spyttkjertler.
Histologisk påvisning av amyloid ved farging med Kongorødt og mikroskopi i polarisert lys er gullstandarden for diagnostisering. Nærmere karakterisering kan gjøres ved immunhistokjemi eller analyse av amyloid ekstrahert fra affisert vev.
Differensialdiagnoser
Det er mange tilstander som kan ligne på amyloidose. En grundig utredning er derfor avgjørende for å stille riktig diagnose.
- Artritt (amyloide skulderputer): Kjennetegnes ved leddsmerter, hevelse og stivhet og kan i sjeldne tilfeller gi opphav til amyloide avleiringer i skulderputene.
- Diabetes mellitus (diabetisk hånd, nefropati): Kan føre til en rekke komplikasjoner, inkludert nevropati (diabetisk hånd) og nyreskade (nefropati) som begge kan ligne på amyloidose.
- Glomerulonefritt (proteinuri): Inflammasjon i nyrenes filtrasjonsenheter som kan føre til proteinuri (protein i urinen), et funn som også kan sees ved amyloidose.
- Inflammatorisk tarmsykdom (IBD): En gruppe kroniske betennelsessykdommer i tarmen som i sjeldne tilfeller kan være assosiert med amyloidose.
- Hjertesykdom: Hjertesvikt, kardiomyopati (hjertemuskel sykdom) og perikarditt (betennelse i hjerteposen) kan gi symptomer som ligner på amyloidose i hjertet som for eksempel kortpustethet, tretthet og hevelse i bena. Restriktiv kardiomyopati: En hjertesykdom som kjennetegnes ved stive hjertevegger som kan ligne på restriktiv kardiomyopati forårsaket av amyloidose.
- Hypotyreose (myksødem): Kan gi opphav til myksødem (hevelse i huden), et funn som også kan sees ved amyloidose.
- Macroglossi av annen årsak (genetisk, hypotyreose, lysosomal lagringssykdom): Forstørret tunge kan skyldes flere tilstander, inkludert genetiske faktorer, hypotyreose og lysosomale lagringssykdommer og kan ligne på macroglossi ved amyloidose.
- Nevrologiske sykdommer: Polynevropati, karpaltunnelsyndrom og andre nevrologiske tilstander kan gi symptomer som ligner på amyloidose i nervesystemet som for eksempel nummenhet, prikking og muskelsvakhet.
- Systemisk sklerose (puffy hands/fingres): Fører til fortykning av huden og indre organer, og som i tidlige stadier kan gi hevelse i hender og fingre (puffy hands) som kan ligne på amyloidose.
- Sklerødem (scleredema): En sjelden tilstand som kjennetegnes ved fortykning av huden, hovedsakelig på overkroppen og som kan ligne på noen former for amyloidose.
- Sjøgrens syndrom (sicca-fenomen, hovne kjertler); Angriper fuktproduserende kjertler som kan gi tørre øyne og munn (sicca-fenomen) samt hovne kjertler. Kan dermed ligne på manifestasjoner av amyloidose.
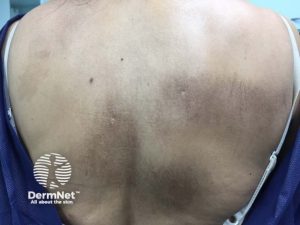
Det finnes ingen kurativ behandling, men symptomene kan lindres med kløstillende kremer og lysbehandling. Noen opplever at flekkene blekner over tid, mens de hos andre er stabile. Illustrasjon: Dermnet. CC BY-NC-ND 3.0 NZ
Behandling
Behandlingen av amyloidose avhenger av den underliggende årsaken, typen amyloidose og pasientens tilstand.
Ved AA amyloidose er et av hovedmålene å behandle den underliggende inflammatoriske sykdommen for å redusere risikoen for forverring og amyloidavleiring. Dette reduserer leverens produksjon av serum amyloid A (SAA), da høye nivåer av SAA øker risikoen for amyloidavleiring. Blant immundempende legemidler har IL-6 hemmeren tocilizumab vist seg effektiv ved ulike manifestasjoner av AA-amyloidose (Inoue D, 2010; Redondo-Pachón MD, 2013).
Ved Familiær Middelhavsfeber, som ofte er årsak til AA-amyloidose, er kolkisinbehandling essensielt i hele sykdomsforløpet som forebyggende tiltak. Dersom sykdommen ikke er godt kontrollert, kan behandling med IL-1-hemmere som anakinra (Kineret) eller canakinumab (Ilaris) forsøkes. Tofacitinib (JAK-hemmer) har også vist lovende resultater i studier (Migita K, 2014).
Ved AL amyloidose vurderes først om pasienten er egnet for autolog stamcelletransplantasjon. Dette er aktuelt for yngre pasienter med god hjerte-, nyre- og lungefunksjon. Omtrent 20% av pasientene med AL-amyloidose tilbys denne behandlingen (Al Hamed R, 2021). For nærmere informasjon om behandling av AL-amyloidose og andre typer amyloidose, se relevant litteratur (Ihne S, 2020).
De siste årene har det vært intensiv forskning på utvikling av medikamenter som kan stanse og reversere amyloidavleiring (Nuvolone M, 2017; Al Hamed R, 2021). Tafamidis (Vyndaqel) har vist seg å bedre overlevelse ved transthyretin-mediert kardial amyloidose, men et endelig gjennombrudd for de fleste amyloidosepasienter gjenstår (Lamb EN, 2021).
Prognose
Prognosen ved amyloidose varierer avhengig av type og antall affiserte organer. Hjerteaffeksjon er den alvorligste manifestasjonen, og ved AL-amyloidose er 40% av dødsfallene forårsaket av hjertesvikt.
Litteratur
Holt MF, 2023 (kardial amyloidose)
