BARN MED REVMATISK SYKDOM. BARNEREVMATOLOGI (REV 053-062)
118 Autoinflammatoriske sykdommer, Periodiske febersyndromer. CAPS, CRMO, Familiær middelhavsfeber, FMF, PFAPA, TRAPS (REV 076)
Jan Tore Gran and Øyvind Palm
Kjennetegn på autoinflammatoriske sykdommer
Små barn med gjentatte (regelmessige eller uregelmessige) episoder med høy feber uten påvisbar infeksjon
Familiære tilfeller
Varierende affeksjon med halssmerter, eksantem, konjunktivitt, artritt og/eller serositt
Høy CRP under anfall, ingen antistoff (ANA, CCP/ACPA er negative)
Gentestening er mulig for flere av tilstandene
ICD-10 M04.9 (uspesifisert autoinflammatorisk sykdom)
Prosedyrekoder: Leddpunksjon/artrocentese (klikk for å spesifisere ledd:) TN_10. UL veiledet leddpunksjon: NXA10K. Intravenøs infusjon: WBGM00.
ATC koder: Anakinra : L04AC03. Canakinumab: L04AC08. Tocilizumab: L04AC07
Kunnskap om autoinflammatoriske sykdommer er en viktig del av kompetansen til en revmatolog. Denne kunnskapen er avgjørende for å gi optimal behandling til pasienter med både revmatiske sykdommer og autoinflammatoriske sykdommer, for å forebygge komplikasjoner og for å forbedre kommunikasjon og samarbeid med andre spesialister. I tillegg kan symptomene ligne på klassiske revmatiske sykdommer, noe som kan gjøre diagnosen vanskelig. Revmatologer med kunnskap om autoinflammatoriske sykdommer er bedre rustet til å skille mellom disse to tilstandene.
Definisjon
Autoinflammatoriske sykdommer omfatter en gruppe genetisk betingede sykdommer som karakteriseres av tilbakevendende feber og ulike organmanifestasjoner, ofte omtalt som periodiske febersyndromer. Disse skyldes ofte en medfødt defekt i ett gen og aktivering av det inate immunsystemet. Symptomer debuterer typisk i tidlig barnealder. Genom sekvensering har de senere år avdekket en rekke nye autoinflammatoriske sykdommer (Miner JJ, 2023).
I tillegg ses andre autoinflammatoriske sykdommer som kan skyldes ervervet mutasjon og begynner senere i livet, slik som ved det nylig beskrevne VEXAS syndrom. Typisk for autoinflammatoriske sykdommer er fravær av auto-antistoffer, relasjon til HLA-gener, og tilstandene ses omtrent like ofte hos begge kjønn.
De autoinflammatoriske sykdommene skiller seg patogenetisk fra autoimmune sykdommer (Feist E, 2018), men en viss patogenetisk overlapp kan foreligge. Ved flere revmatiske sykdommer foreligger dessuten en kombinasjon av autoinflammasjon og autoimmunitet i patogenesen. Det gleder blant annet for ankyloserende spondylitt/Bekhterevs sykdom, arteritis temporalis og Behcets sykdom. Disse betegnes likevel ikke som autoinflammatoriske sykdommer vanligvis (Doria A, 2012; Szekanecz Z, 2021).
Etiologi og patogenese
Autoinflammatoriske sykdommer kan inndeles etter gen-defekt eller feil i patogenesen:
- MEFV-genet: familiær middelhavsfeber (FMF)
- NLRP3-genet: Cryopyrin assosierte febersyndromer (CAPS)
- Inflammosomopatier: FMF, FCAS, NLRC4 relatert MAS NAIAD
- Proteasom-assosierte sykdommer: PRAAS, CANDLE
- Interferonopatier: AGS, SAVI, Singleton-Mertens syndrom, COPA
- NF-κB signalvei (ubiquitinering): HA20, HOIL2-mangel, haploinsuffisiens av RELA, CRIA, VEXAS.
De fleste periodiske febersyndromer skyldes mutasjon i ett enkelt gen. Unntaket er PFAFA (se nedenfor) som antas være multigenetisk betinget. Sykdommene medfører et dysfunksjonelt immunsystem som utløser eksessiv systemisk inflammasjon via det innate / medfødte immunsystemet og debuterer typisk tidlig i livet.
Genetiske tester: For informasjon om aktuelle gen-tester er genetikkportalen.no aktuell.
Historie
Begrepet “autoinflammasjon” har fått økt betydning innen revmatologi siden 1999, da tilstanden ble definert som “episoder med tilsynelatende uprovosert inflammasjon i fravær av autoantistoffer eller autoreaktive T-celler” (McDermott MF & Kastner D, 1999).
Denne definisjonen understreker at autoinflammatoriske sykdommer skiller seg fra autoimmune sykdommer, der immunsystemet feilaktig angriper kroppens egne vev. Ved autoinflammasjon er det en dysregulering av det medfødte immunsystemet som fører til ukontrollert betennelse, uten at det nødvendigvis foreligger spesifikke autoantistoffer eller autoreaktive T-celler.
Initialt ble sykdommene definert som utelukkende interleukin-1 relaterte, noe som endret seg fra 2009 da sammenhenger også med andre cytokiner ble beskrevet (Masters SL, 2009).
Symptomer
Typiske symptomer på autoinflammatoriske sykdommene inkluderer:
- Feber: Høy feber, ofte over 38°C, er et vanlig symptom. Feberen kan være periodisk eller vedvarende.
- Nattesvette: Overdreven svetting om natten er et typisk symptom.
- Smerte og betennelse: Smerter og betennelse i ledd, muskler og andre vev kan forekomme.
- Hudutslett: Ulike typer hudutslett, som erythem og eksem (kløende utslett), kan være til stede.
- Munnsår: Sår og betennelse i munnslimhinnen (stomatitt) kan oppstå.
- Serositt: Perikarditt, pleuritt eller peritonitt kan forekomme.
- Artritt: Betennelse i leddene kan føre til smerte, stivhet og hevelse.
- Affeksjon av andre organer: I noen tilfeller kan sykdommene også ramme andre organer, som milten, leveren, lymfesystemet eller nervesystemet.
Utbrudd kan provoseres frem av traumer, kirurgi, kulde og infeksjoner.
Utbredelse og debutalder
autoinflammatoriske sykdommer rammer både menn og kvinner. Selv om sykdommene kan debutere i alle aldre, er barn oftere rammet, med unntak av FMF, PAPA, Adult Stills, Schnitzlers syndrom, VEXAS.
| Forskjeller på autoinflammasjon og autoimmune sykdommer (Krainer J, 2020) | ||
| Autoinflammasjon | Autoimmunitet | |
| Immunologisk dysregulering | Innate immunsystem | Adaptive immunsystemet |
| Predominante celletyper | Monocytter, makrofager, neutrofile | T-celler, B-celler |
| Cytokiner som er mål for behandling | TNF, IFNαβ, IL-1, IL-2, IL-12, IL-23, IL-18 | IFNγ, TNFα, IL-1, IL-2, IL-4, IL-6, IL-5, IL-9, IL-10, IL-12, IL-13, IL-17, IL-22, IL-23 |
| Patogenese for organskade | Neutrofil- og makrofagmediert | Autoantistoff- eller autoantigen-spesifikk T-celle-mediert |
Undersøkelser
Anamnesen omfatter alder ved debut, forløpet (episoder, varighet av intervall eller vedvarende), aktuelle symptomer. Er feber målt (metode) og er det laget en feberkurve?
Klinisk ses nedsatt allmenntilstanden med feber, utmattelse og smerter er typisk ved anfall og helt normale forhold i mellomtiden ved periodiske febersyndromer, mens vedvarende inflammasjonstegn kan prege de øvrige autoinflammatoriske sykdommene.
Laboratorieundersøkelser omfatter inflammasjonsprøver (SR og CRP) som forventes å være tydelig forhøyet ved sykdomsaktivitet. Også antall trombocytter og leukocytter og ferritin kan øke. Andre aktuelle prøver til dels av differensialdiagnostiske årsaker omfatter elektrolytter, lever-, nyre-, og thyreoidea-funksjonsprøver, D-dimer, ferritin, fibrinogen, triglyserid, glukose og kreatin kinase CK. Løselig IL- reseptor, ANA og antifosfolipid antistoff (lupus antikoagulant, anti-kardiolipin- og beta-2 glykoprotein) også være aktuelt. Urin-stiks. Gentester er ofte avgjørende for diagnosen; se genetikkprotalen.no.
Bildediagnostikk. Initialt tas ofte røntgen eller CT av lunger for å utelukke infiltrat og tegn til pleuravæske. Ved ultralyd kan patologiske tegn i nyrer, lever, lymfeknuter eller milt påvises. Ellers rettes undersøkelsene seg mot aktuelle symptomer og kliniske funn.
Ekkokardiografi kan være aktuelt med tanke på endokarditt, perikarditt eller klaffepatologi.
Behandling
Et tegn er også at autoinflammatoriske sykdommer responderer relativt dårlig på klassisk anti-inflammatorisk behandling med kortikosteroider og csDMARDs.
Mange av de autoinflammatoriske sykdommene, særlig de periodiske febersyndromene kjennetegnes ved dysregulering av IL-1 signalering i immunsystemet. De kan behandles effektivt med biologiske legemidler i form av IL-hemmere som anakinra (Kineret) eller canakinumab (Ilaris), men det er unntak. TNF-hemmeren etanercept og IL-6 hemmeren tocilizumab brukes i noen tilfeller. For uspesifikke eller ervervede autoinflammatoriske sykdommer brukes i større grad kortikosteroider og klassiske DMARDS (Krainer J, 2020).
Nedenfor er en del av de hittil kjente autoinflammatoriske sykdommene listet:
A20 haploinsuffisiens (HA20), TNFAIP3 mutasjon
A20 Haploinsuffisiens er en monogen autoinflammatorisk sykdom med et Morbus Behcets-lignende forløp, første gang beskrevet i 2016 (Zhou Q, 2016).
Sykdomsårsak: Heterozygot arvelig mutasjon i TNFAIP3 som koder for NF-κΒ regulerende protein A20. Lav A20 funksjon øker produksjonen av proinflammatoriske cytokiner som IL-1, IL-6 og TNF-α (Zhou Q, 2016).
Debutalder: Vanligvis barn før 10 års alder, men kan debutere fra spedbarnsalder til unge voksne.
Symptomer: Residiverende smertefulle orale sår med ulcerasjoner er vanligst, men også tilsvarende sår videre nedover i gastrointestinal-tractus er vanlig, og genitale sår forekommer. Abdominal smerte og i verste fall perforasjoner er sett. De fleste har polyartritt, noen bare artralgi. Hudlesjoner kan omfatte pustler, follikulitt, patergi (hudreaksjon på stikk/skade), akne og abscesser. Inflammasjon i øyne av ulike typer som anterior uveitt/iridocyklitt og retinal vaskulitt.
Laboratorieprøver: Høye inflammasjonsparametere som CRP og SR. Hos noen er ANA og anti-DNA forhøyet.
Genetikk: Alle har mutasjon i TNFAIP3 genet. En assosiasjon med HLA-B51 som ved Behcets sykdom foreligger ikke.
Diagnose: Ved typisk anamnese og kliniske funn er genetisk undersøkelse avgjørende.
Differensialdiagnoser: Behcets sykdom, juvenil lupus (JSLE), JIA, PFAPA (se nedenfor), inflammatorisk tarmsykdom (IBD).
Behandling: Biologiske legemidler i form av IL-1 hemmere (anakinra, canakinumab) eller TNF-hemmere har effekt hos de fleste.
Litteratur: Bitter H, 2023, Zhou Q, 2016; Aeschlimann AF, 2018
Behcets sykdom
Sykdommen klassifiseres ofte som vaskulitt., men en assosiasjon med autoinflammatorisk patogenese er sett. Debut oftest i 20-30 årene, fravær av antistoff og forverring som delvis er relatert til traumer (patergi) kjennetegner sykdommen. Respons fra det innate immunsystemet med økt produksjon av pro-inflammatoriske komponenter som TLR, neutrofile NK-celler eller γδ T-celler (van der Houwen TB, 2022). Mindre typisk for autoinflammatorisk sykdom er assosiasjonen med HLA B51. Vennligst les mer om Behcets i eget kapittel.
Blau syndrom
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Pediatrisk granulomatøs artritt (familiære tilfeller kalles Blau syndrom, mens sporadiske benevnes neonatal sarkoidose)
Sykdomsårsak: Sykdommen skyldes mutasjoner i “caspase recruitment domain” (CARD 15) som også kalles NOD2 genet.
Debutalder: 2-4 år er vanligst
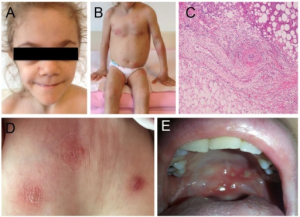
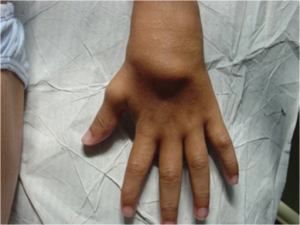
Symptomer: Triade av granulomatøs uveitt (ofte bilateral), artritt og utslett (brunaktige subkutane plakk, lichenoid-liknende). Leddsmerter, evt. synoviale cyster som kan føre til deformiteter. Erythematøst makulopapulært utslett og små subkutane gul-brune knuter som kan flasse (lichenoid) på overflaten. Histologisk ses granulomer med kjempeceller. Symmetrisk polyartritt kan omfatte håndledd, MCP, PIP, MTP1, ankler og albuer. Typisk utvikles kontrakturer i tidlig sykdomsfase (camptodaktyli) (Wouters CH, 2014). I tillegg medfører granulomatøs inflammasjon at periartikulære strukturer og sener kan hovne betydelig opp, særlig over håndrygg og håndledd (illustrasjonen) (Pac Kisaarslan PA, 2020). Illustrasjon: Wouters CH, 2014. CC BY 4.0. Illustrasjon: Hu J, Chin Med J, 2017. CC BY-SA 3.0.
Bildediagnostikk; Erosjoner ikke beskrevet, men håndledds-ankylose forekommer.
Biopsi: Blau syndrom er den eneste autoinflammatoriske sykdommen som danner granulomer. En påviser epiteloide ikke-nekrotiserende granulomer med flerkjernede kjempeceller som ved sarkoidose (early-onset sarcoidosis).
Genetikk: Alle har mutasjoner i NOD2 genet, men ulik penetrans gjør at enkelte er asymptomatiske.
Differensialdiagnoser: Tuberkulose, Juvenil idiopatisk artritt (JIA), juvenil systemisk lupus (jSLE), Crohns sykdom, familiær middelhavsfeber (FMF), Vogt-Koyanagi-Harada syndrom (VKH):
Behandling: Behandles med IL-1 hemmere, TNFα- blokkere, sjeldnere med thalidomid.
Litteratur: Punzi L 2009; Wouters CL, 2014
CANDLE
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Kronisk atypisk neutrofil dermatose med elevert temperatur. CANDLE = Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature
Sykdomsårsak: Sykdommen skyldes mutasjon av PMSMB8-genet (proteasome subunit beta type-8). Proteasomer er store proteinkomplekser i cytoplasma og cellekjernen. Deres funksjoner er å degradere unødige eller ødelagte proteiner. Proteasomer destruerer proteiner som er merket med et lite protein som kalles ubiquitin.
Forekomst: Starter i barnets første leveår, ofte de 2-4 første leveukene.
Symptomer: Initialt sees anfall av daglig eller neste daglig feber. Utslett (erythematøst, annulært) som varer noen få dager eller uker. Senere utvikles periorbitalt erythem og ødem (differensialdiagnose: juvenil dermatomyositt), hevelse i fingre og tær, hepatomegali, nedsatt trivsel og utvikling, lipodystrofi og lymfadenopati. Sjeldnere manifestasjoner er perioral hevelse, parotitt, konjunktivitt/episkleritt, acanthosis nigricans, hypertrikose, artralgi, pannikulitt og alopecia areata.
Hudbiopsi viser tette interstitielle infiltrater av mononuklære celler.
Genetikk. Variable kombinasjoner mutasjoner. Oftest er pasientene homozygote eller delvis heterozygote for PSMB8 mutasjoner, men andre forekommer også.
Differensialdiagnoser: Neonatal-onset multisystem inflammatory disease (NOMID), Cryopyrin-associated periodic syndromes (CAPS), Familiær middelhavsfeber (FMF), TRAPS, Juvenil idiopatisk artritt (JIA), Kawasaki sykdom, Sepsis.
Behandling: Dessverre har en ikke funnet spesielt effektiv spesifikk terapi. Behandlingsmessig kan forsøkes med perorale kortikosteroider og metotreksat. Ved utilfredsstillende effekt responderer noen på IL-1 reseptor antagonister, TNFα-blokkere og IL-6 blokkere (Liu Y, 2011).
Litteratur: Torrelo A, 2017
CAPS (Cryopyrin assosierte febersyndromer)
ICD-10: M 04.2
CAPS omfatter tre tilstander som skyldes mutasjon i samme gen: FCAS, Muckle-Wells syndrom og NOMID/CINCA
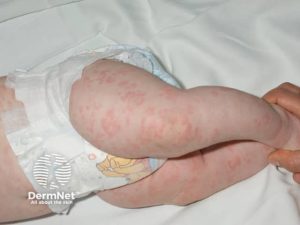
Sykdomsårsak: Mutasjon i NLRP3-genet som koder for cryopyrin. NLRP3 er en komponent av et multiprotein kompleks som kalles inflammasom som har til funksjon å aktivere IL-1B (“inflammasomopati”). Komplekset gjenkjenner NLRP3 gjennom sitt LRR (C-terminal leusin-rich repeats) domene til ulike mikrobielle substanser og endogene faresignaler. Inflammasomer er reseptorer i cellenes cytosol som regulerer aktiviteten av kaspase-1 som så påvirker omdannelsen av pro-1L-1B til aktiv IL-1B. Vanligvis kreves to signaler for å redusere produksjon av IL-1B. Ved de fleste autoinflammatoriske sykdommene er avhengigheten av signal 2 mindre. Av og til er imidlertid produksjon av IL-B ikke vesentlig høyere enn normalt, men derimot raskere. Produksjonen av IL-1Ra (reseptor) antagonist er lavere. Dette er grunnen til at IL-1 hemmere (anakira, canakinumab) ofte benyttes i behandlingen.
Prevalens: 1-3/million
Debutalder: Median alder ved debut er funnet å være 0,8 år, men med diagnose først ved 15 års alder er vanlig
Symptomer: Feber, urtikarielt utslett, røde øyne og artralgi. Varighet 1-2 dager. Nevrologiske manifestasjoner omfatter hodepine, nevralt hørselstap og nervus opticus-affeksjon.
Genetikk. Testing av NLRP3 genet
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, Juvenil idiopatisk artritt (JIA), Systemisk lupus erythematosus (SLE), tilbakevendende virusinfeksjoner, kulde-urtikaria.
Behandling: Ved cryopyrin-assosierte syndromer kan biologiske legemidler i form av IL-1 blokkere med lang halveringstid forsøkes: canakinumab (Ilaris) 150 mg sub-kutant hver 8. uke. Anakinra (Kineret) kan også gi effekt, men sentralnervøs sykdom kan kreve høye doser (3-10 mg/kg/d).
Litteratur: Keddi S, 2018
CAPS/ Familiær kulde autoinflammatorisk syndrom (FCAS)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Neonatal onset multisystem inflammatorisk sykdom (NOMID/CONCA).
Symptomer: Konjunktivitt. Anfall etter eksposisjon for kulde.
CAPS/MWS: Muckle-Wells syndrom: Vennligst se Muckle-Wells syndrom nedenfor
CAPS/NOMID/CINCA: Vennligst se CINCA nedenfor
CINCA: Kronisk infantil nevrologisk kutant og artikulært syndrom (CINCA)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: NLRP3 mutasjon. Autosomal dominant genetisk sykdom. Genet koder for nøkkelkomponenter for regulering av aktivering og sekresjon av pro-inflammatorisk interleukin (IL)-1β. Imidlertid mangler 30-40% denne mutasjonen. Andre former omfatter mutasjoner i NLRP12 genet (vennligst se NLRP12 assosiert febersyndrom nedenfor).
CINCA er en undergruppe cryopyrin assosiert febersyndrom (CAPS) med alvorlig forløp, vennligst se ovenfor. Muckle Wells syndrom (se nedenfor) er en mildere variant av CAPS
Prevalens: 12/million
Debutalder: De fleste debuterer allerede de første dagene etter fødselen med et kronisk urtikarielt utslett, lavgradig feber og persisterende forhøyede inflammasjonsparametere.
Symptomer: Feber, utslett, uveitt, kronisk meningitt, cerebral atrofi, forsinket utvikling, ev. papilleødem. Karakteristisk ansiktsform med prominent panne forekommer.
Genetikk: Påvisning av NLRP3 mutasjoner.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer, juvenil idiopatisk artritt (JIA), Juvenil systemisk lupus erythematosus (jSLE), kongenitt infeksjon (som er tilstede ved fødselen), som for eksempel cytomegalovirus (CMV) eller toksoplasmose. Meningitt. Histiocytose.
Behandling: IL-1 hemmer: Anakinra, canakinumab.
Litteratur: Finetti M, 2016
CRMO (chronic multifocal osteomyelitis), CNO

Synonym: Chronic nonbacterial osteomyelitis (CNO). Kronisk tilbakevendende multifokal osteomyelitt begynner oftest i barnealderen og blir ofte feiltolket som infeksjon. Hos voksne klassifiseres tilstanden oftere som en undergruppe av SAPHO syndromet.
Debutalder: Barn etter 2 års alder. De fleste er mellom 8 og 13 år. Median alder ved diagnose er 10 år.
Epidemiologi: Sjelden sykdom, men økende oppmerksomhet og bedre diagnostikk (MR) medfører at flere tilfeller er diagnostisert i senere tid.
Symptomer: Smerter i skjelett eller ledd med eller uten relatert hevelse. Mest symptomer om natten eller morgenen kan redusere nattesøvnen. Noen debuterer med patologisk fraktur. Hudforandringer med akne eller pustulose er sjelden hos barn. Artritt hos ca. 30%.
Laboratorieprøver: Normale eller lett økt CRP og SR. Andre prøver som sjekkes er celletellinger (hvite, trombocytter og hemoglobin/erytrocytter).
Bildediagnostikk: Ved konvensjonell røntgen eller CT kan osteolytiske eller cystiske lesjoner ses. Vanligste lokaliseringer er metafyser i lange rørknokler, bekken og vertebra. Også kjeveben kan affiseres, mens hodeskallen angripes nesten aldri. MR-undersøkelse er imidlertid mer sensitiv fordi også tegn til inflammasjon med ødem vises. For å avdekke omfanget av multifokal utbredelse er helkropps-MR aktuelt. Dersom en vil utrede lokaliserte symptomer, gir derimot MR av aktuelt område best bilder og mest informasjon.
Genetikk: Heterozygot mutasjon i det filamin-bindende domenet påFBLIM1 genet er påvist i noen tilfeller. Mutasjonen medfører økte mengder pro-inflammatoriske cytokiner som IL-1β, IL-6, og TNFα.
Differensialdiagnoser: CRMO er en eksklusjonsdiagnose. I uklare tilfeller er biopsi fra skjelett-manifestasjoner aktuelt for å utelukke kronisk infeksjon, malignitet eller annen systemsykdom. Viktige differensialdiagnoser er leukemi, lymfom, primære og sekundære skjelett-tumorer. Infeksjoner omfatter bakteriell osteomyelitt og tuberkulose med eller uten samtidig immunsvikt. Langerhans histiocytose, PAPA, DIRA (se nedenfor) og Majeed syndrom (mutasjon i LIPIN2 genet) er sjeldne tilstander som også ligner CRMO.
Behandling. Initialt benyttes NSAIDs som ofte er tilstrekkelig. Hos noen er også kortikosteroider, DMARDs (metotreksat eller sulfasalazin), TNF-hemmere eller bisfosfonater.
Forløp: Prognosen på lang sikt er god, selv om de fleste opplever flere residiv. Remisjonsraten er 40% i løpet av 1-5 år, men tilbakefall etter opp til 15 år er observert. Komplikasjoner i forløpet ses hos en av tre og omfatter vertebrale frakturer, skjelett deformiteter og kronisk smerte.
Illustrasjon: Cox AJ PLoS One, 2017. CC BY 4.0.
Litteratur: Johnsson A, 2015; Hofmann SR, 2017; Koryllou A, 2021.
DEX (defiency in ELF4, X-linked)
Gutter med inflammasjon i slimhinner, orale ulcera feber og abdominale smerter og diare.
Litteratur: Tyler M, 2021
DIRA (deficiency of the IL-1R antagonist)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Mutation i IL1RN genet som koder for interleukin-1 (IL-1) reseptor antagonist (IL-1Ra). IL-1Ra mangel medfører in høy inflammatorisk aktivitet via IL-1 og IL-1 reseptor (IL-1R1).
Forekomst: Debut mellom de første dager etter fødsel og innen få uker.
Symptomer: Eksem og multifokale osteolytiske lesjoner (ligner septisk osteomyelitt), periostitt, pustulose, evt. heterotrofisk bennydannelse i proksimale femur, trombose og en sjelden gang vaskulitt. Vedvarende forhøyede inflammasjonsparametere. Kronisk lungesykdom og tromboser rapportert (Jesus AA, 2011).
Genetikk. Diagnosen sikres ved påvisning av mutasjoner i IL1RN genet.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer, Juvenil idiopatisk artritt (JIA), spesielt systemisk JIA, kongenitte infeksjoner (cytomegalovirus (CMV), toxoplasmose), epidermolysis bullosa, Kostmann syndrom.
Behandling: IL-1 hemmer (anakinra, canakinumab) er ofte effektiv og viktig. Ved intoleranse, kan hyposensibilisering mot medikamentet være aktuelt.
Litteratur: Mendonca LO, 2017
DITRA (Deficiency of interleukin-36 receptor antagonist)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Mutasjoner i IL36RN genet medfører mangel på IL-36 reseptor antagonist
Alder ved debut: Median 7 måneder
Symptomer: Feber og generalisert pustuløs palmoplantaris. Gutter angripes oftest.
Differensialdiagnoser: Pustuløs psoriasis (GPP), plakkpsoriasis, akutt generalisert eksantematøs pustulose (AGEP), subkorneal pustuløs dermatose (Sneddon-Wilkinson sykdom), infeksiøs pustulose, andre autoinflammatoriske sykdommer (CAPS, PAPASH (Pyogen artritt, pyoderma gangrenosum, akne og suppurativ hidradenitt), medikamentreaksjoner.
Behandling: kortikosteroider, DMARDs, biologiske legemidler: TNF-hemmer og IL-1 hemmere er brukt
Litteratur: Hospach C, 2019
Disabling pansclerotic morphea (DPM)
DPM er en sjelden, systemisk autoinflammatorisk sykdom som kjennetegnes ved redusert sårtilhelning, fibrose, cytopenier, hypogammaglobulinemi og økt forekomst av SCC (Squamous celle carcinom). Sykdommen skyldes mutasjon i STAT4-genet (Baghdassarian H, 2023).
Early onseth enterocolitis (mutasjoner i IL-10R).
Sykdomsårsak. Homozygot mutasjoner i genene IL10RA ogIL10RB. Mutasjonene medfører økt sekresjon av TNF-α og andre proinflammatoriske cytokiner.
Symptomer og debutalder: Alvorlig, enterokolitt som begynner i barnealder. Blodig diare, kolon-abscesser, perianale fistler, orale ulcera.
Differensialdiagnoser: Inflammatoriske tarmsykdommer, infeksiøs gastroenteritt, clostridium difficile infeksjon, melkeproteinallergi, cøliaki, IPEX syndrom (Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome)
Behandling: Ved manglende effekt av immunsuppressive medikamenter, er allogen stamcelle transplantasjon en mulighet
Litteratur: Glocker E-O, 2009
Familiær Middelhavsfeber (FMF)
ICD-10 E85.0 (familiær amyloidose)
“Recurrent Hereditary Polyserositis”
Sykdomsårsak: Autosomal recessiv. Mutasjon i MEFV genet på kromosom 16. Genet koder for pyrin som kan danne pyrininflammasjoner (“inflammasomopati”) som kan aktivere interleukin 1-beta og medføre systemisk inflammasjon. Genetiske variasjoner er vanlig.
Debutalder: Fire av 5 er under 20 år, to av tre under fem år v ed første symptom. Debut etter 30-års alder er uvanlig.
Forekomst: Den vanligste monogene autoinflammatoriske sykdommen. Mer enn 100,000 er angrepet på verdensbasis . FMF rammer først og fremst Non-Askenazi jøder, armenere, tyrkere, arabere og italienere. En studie fra Danmark viste samlet prevalens på 1/11.680 personer (0,009%), omtrent likt fordelt mellom kvinner og menn. De fleste (41,8%) var av tyrkisk avstamning, etterfulgt av libanesere (15,8%), og syrere (6,5%). De øvrige var fra Asia og Nord-Afrika. Bare 0,8% var av Vest-Europeisk avstamning (Mortensen SB, 2020).
Symptomer: Typisk triade er residiverende episoder med feber, abdominal smerte og artritt i store ledd. Oftest mangler forutgående utløsende årsak. Varigheten er 1-3 dager. Forut for anfall kjenner de fleste pasientene prodromer. De aller fleste merker utmattelse. Forutfor peritonitt som er vanligste symptom oppstår kvalme hos 37%, appetitt-løshet hos 30%. Tilsvarende, før pleuritt (nest vanligste symptom, ofte unilaterale thoraks-smerter) merkes dyspne (34%, utstrålende ubehag i ryggen (17%) og hoste (13%), før artritt har 45% nummenhet og 26% leddsmerter. Erysipelas-lignende erythem er mer sjelden, men forutgås av en brennende følelse hos 64% (Babaoglu H, 2020). Anfall av 1-3 dagers varighet, 2-4 ukers intervaller, monartritt (store ledd), Obs! kronisk coxartritt, sakroiliitt, treningsutløst myalgi, erythem på legger (likner erysipelas), serositt (peritonitt 90%, pleuritt 45%, perikarditt 1 %), splenomegali. Henoch-Schönleins purpura / IgA vaskulitt, PAN liknende hudforandringer er mulig. Kan debutere med bare feber. Asymptomatiske utenfor anfall.
Amyloidose er en fryktet komplikasjon etter flere års sykdomsforløp. Symptomer er progredierende nevropati, proteinuri og nyresvikt. Konsekvent behandling av FMF forhindrer utvikling av amyloidose.
Svangerskap: Kommentar til tabell nedenfor: Relativt høy andel med komplikasjoner og oppbluss under svangerskapet kan skyldes en lav andel som brukte kolkisin.
| Svangerskap og fødsel blant 979 pasienter i Tyrkia (Bodur H, 2020) | Antall (%) |
| Friske levende fødte | 396 (67,7) |
| Tidligfødsler | 41 (7,0) |
| Spontanaborter | 106 (18,1) |
| Dødfødsler | 32 (5,5) |
| Fetale malformasjoner | 10 (1,7) |
| Forløp i svangerskapet
-Oppbluss i svangerskapet |
154 (38,4) |
| -Kraftigere episoder enn før svangerskap | 54 (13,5) |
| -Kolkisin under svangerskapet | 62 (15,4) |
| -Lavere kolkisin-dose under svangerskapet | 28 |
| -Uendret kolkisin-dose | 32 |
| -Høyere kolkisin-dose | 2 |
| -Antall episoder med sykdomsoppbluss i svangerskapet (gjennomsnitt) | 4,94 |
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, Juvenil idiopatisk artritt (JIA), spesielt systemisk JIA, inflammatorisk tarmsykdom (IBD), juvenil systemisk lupus erythematosus (jSLE), tilbakevendende virusinfeksjoner, tuberkulose, akutt intermittent porfyri.
Behandling: Kolkisin er generelt trygt og fordelaktig å kontinuere i svangerskap (Ben-Chetrit E, 2010). Doseforslag kan være :Voksne 1,2-1,8 mg. Anfall: 0,6 mg/time i 4 timer, deretter hver annen time første dag. Barn < 5 år: Startdose maks 1,0 mg. > 10 år: Startdose maks 1,5 mg. Noen får bivirkninger som omfatter diare, abdominal smerte, utslett, leukopeni, trombocytopeni, nevropati, myopati og leverenzymstigning. IL-1 hemmer (anakinra, canakinumab) kan brukes ved non-respons eller intoleranse. Alternativer er TNF-alfa hemmer eller IL-6 hemmer (tocilizumab). Behandlingen skal kontinueres i mange år.
Litteratur: Kucuk A, 2014
Hyper IgD syndrom (HIDS)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Autosomal recessiv. Mutasjon i MVK genet (mevalonat kinase). Alvorlig enzymmangel gir Mevalonsyre aciduri.
Forekomst: Hyppig i Nord-Europa, særlig hos franskmenn og nederlendere. Anfall over 5-7 dager med 4-8 ukers intervaller. Anfallene kan utløses av vaksinasjoner eller infeksjoner.
Debutalder: Småbarn.
Symptomer: Feber, utslett, artralgi, artritt (70 % non-erosiv og gjerne polyartikulær, symmetrisk og transiente), abdominalsmerter (ofte uttalte med oppkast og diare), lymfadenopati (cervikalt), hodepine, splenomegali, orale og genitale after. Alvorlige former også med nevrologiske-, synstap og hørselsutfall.
Laboratorieprøver: Økt IgD hos 78%, evt. også IgA. Høy SR og CRP, samt leukocytose. Anfallene avtar ofte i voksenalder. Amyloidose hos 3 %.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, juvenil idiopatisk artritt (JIA), spesielt systemisk JIA,Inflammatorisk tarmsykdom (IBD), tilbakevendende virusinfeksjoner. Noen metabolske sykdommer, for eksempel mitokondriesykdommer.
Behandling: NSAIDs, steroider, evt. kan TNF-hemmer (etanercept) eller IL-1 hemmer (anakinra, canakinumab) forsøkes.
Litteratur: Steichen et al. J Rheumatol 2009, van der Hilst & Frenkel. J Clin Rheum 2010
JMP Joint contractures – muscle atrophy – microcytic anemia – panniculitis-induced lipodystrophy
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Sannsynligvis samme mutasjon som ved CANDLE syndrom, men JMP debuterer i voksen alder. Proteosom mutasjon.
Debutalder: JMP debuterer i voksen alder
Symptomer: Ledd-kontrakturer, muskelatrofi, microcytær anemi, pannikulitt og lipodystrofi.
Differensialdiagnoser: Muskeldystrofier, kongenitte myopatier, andre genetiske lipodystrofier (Berardinelli-Seip kongenital lipodystrofi, Dunnigan familiær partiell lipodystrofi), pannikulitt med annen årsak. Mikrocytær anemi ved Jernmangelanemi, thalassemi eller sideroblastisk anemi.
Litteratur: Agarwal AK, 2010
Muckle Wells syndrom

ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Dominant autosomal. Muckle Wells syndrom er en undergruppe av cryopyrin-assosiert periodisk syndrome (CAPS) der CINCA er en mer alvorlig form (vennligst se ovenfor).
Prevalens: 1-10 / million, oftest blant kaukasiere.
Debutalder: små barn, median alder 0, 8 år
Symptomer: Episoder med akutt urticaria, konjunktivitt, residiverende feber, artralgi og fatigue er de vanligste symptomene. Oligoartritt, clubbing, episkleritt, hørselstap (70 %), abdominal smerte, migrerende cellulitt, myositt, fasciitt og amyloidose (25 %) ses også. Feberanfall varer 12-48 timer.
Genetikk: Mutasjon i CIA51 genet.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, juvenil idiopatisk artritt (JIA), spesielt systemisk JIA, juvenil systemisk lupus erythematosus (jSLE), tilbakevendende virusinfeksjoner.
Behandling: IL-1 hemmer anakinra, canakinumab. Startes så tidlig i sykdomsforløpet som mulig.
Litteratur: Tran TA, 2017
Nakajo-Nishimuras sykdom (NNS)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Mutasjon i PSMB4 (proteasom). Autosomal recessiv arv, men ca. 50% har ikke lignende i familien.
Debutalder: Oftest omkring 2 års alder, men noen først i 6–12 års alder
Symptomer: Periodisk feber, pernio-liknende hudlesjoner, lipodystrofi særlig i øvre del av kroppen, leddkontrakturer og lange fingre med clubbing. Noen har heliotropt eksem (Differensialdiagnose: Juvenil dermatomyositt). En klinisk overlapp med CANDLE syndrom og delvis genetisk overlapp med JMP-syndrom (joint contractures, muscular atrophy, microcytic anemia, og panniculitis-associated lipodystrophy) forekommer (se ovenfor).
Laboratorium: I blodprøver er CRP og SR alltid forhøyet. CK forhøyet hos noen. Hypergammaglobulin og forhøyet ANA-test (med DNA og SSA-antistoff), også MPO-ANCA forekommer.
Genetikk: Mutasjon i PSMB4 (proteasom).
Differensialdiagnoser: Andre autoinflammatoriske sykdommer, juvenil dermatomyositt, juvenil systemisk lupus erythematosus (jSLE), Kongenitte infeksjoner som for eksempel cytomegalovirus (CMV) eller toxoplasmose. Progeria.
Behandling: Kortikosteroider, DMARDs, biologiske legemidler
Litteratur: Ohmura K, 2019
NLRP12-assosiert syndrom (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak. Syndromet er relatert til CAPS (se ovenfor), men mutasjonen er i NLRP12 genet, ikke NLRP3 . Dominant arv, men variable ekspressiv og penetrans.
Debutalder: Tidlige barneår.
Symptomer: Ligner CAPS (se ovenfor): Høy feber, anfall hver 3.-4. uke, varer 2-10 dager, ofte utløst av kulde, artralgi, myalgi, abdominal smerte og oppkast, evt. aftøse munnsår og lymfadenopati. Symptomer kan også være kuldeindusert.
Genetisk: molekylær screening av NLRP12 gene brukes diagnostisk
Litteratur: Borghini S, 2011
PAPA syndrom (Akne, steril artritt eller abscess, pyoderma gangrenosum)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Skyldes mutasjoner i PSTPIP1 (eller CD2 bindende protein).
Andre autoinflammatoriske syndromer som er forårsaket av mutasjoner i PSTPIP1 genet eller har kliniske overlappende manifestasjoner med PAPA syndromet kan inkluderes i sykdomsgruppen PAPA-lignende sykdommer. Disse omfatter PASH (Pyoderma Gangrenosum [PG], akne og hidradenitis suppurativa), PAPASH (PASH assosiert med pyogen steril artritt), PsAPASH (PASH kombinert med psoriasis artritt, PASS (PG, akne, ankyloserende spondylitt, med- eller uten HS), PAC (PG, akne og ulcerøs kolitt) og PAMI syndromet (PSTPIP1-assosiatert myeloid-relatert-proteinemi inflammatorisk syndrom).
Debutalder: Hud-manifestasjoner begynner blant barn og unge voksne, artritt i løpet av 10-30 års alder.
Symptomer
-Hud kan fremvise patergi fenomen som innebærer at pustler og ulcerasjoner utvikler seg etter små skader. Pyoderma gangrenosum og uttalt akne begynner oftest i tidlige ungdomsår og vedvarer inn i voksen alder. Pyoderma gangrenosum kan utvikle seg til å bli den mest alvorlige manifestasjonen.
-Ledd: Smertefull, residiverende monoartritt utløst av mindre skader. Oftest i albuer, knær og ankler. Symptomene fra ledd er vanskelig å skille fra septisk artritt før leddvæske-analyser avsluttet.
Genetikk: Mutasjoner i PSTPIP1
Differensialdiagnoser: Andre autoinflammatoriske sykdommer, Juvenil idiopatisk artritt (JIA), Inflammatorisk tarmsykdom (IBD), Behcets sykdom, pyoderma gangrenosum med annen årsak, nekrotiserende fasciitt og ekthyma gangrenosum, septisk artritt, impetigo, cellulitt.
Behandling: Ciclosporin A kan ha god effekt på pyoderma gangrenosum ved denne tilstanden (Kanameichi S, 2016). IL-1 blokkere kan forsøkes mot artritt, evt. TNF-hemmer (etanercept). Leddpunksjon og kortikosteroid injeksjoner er aktuelt mot artritt.
Litteratur: Genovese G, 2020
PFAPA (Periodisk feber, aftøs stomatitt, pharyngitt of adenopati syndrom)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
PFAPA er det vanligste periodiske febersyndromet i barnealder (utenom FMF som oftere debuterer noe senere) (Rigante D, 2009). Sykdomsepisodene avtar i intensitet og antall utover i ungdomsårene. Enkelte har symptomer også i voksen alder.
Sykdomsårsak: Patogenesen er uklar, men familiære tilfeller tyder på en genetisk årsak. Blant aktuelle gener er NLRP3 og MEFV, men ingen av disse variantene alene ser ut til å forårsake sykdommen som antas å ha en polygenetisk etiologi. en multigenetisk antas i motsetning til monogenetisk ved de fleste andre autoinflammatoriske sykdommene. En dysregulering av den inate immunsystemet og bakteriekolonier i tonsiller kan være av betydning (Esposito S, 2014). PFAPA syndromet medfører en immunmediert cytokin dysfunksjon (Stojanov S, 2011).
Debutalder: Sykdomsdebut er før 5 års alder. Symptomene går vanligvis helt tilbake innen voksen alder.
Symptomer: Triaden av aftøs stomatitt, cervical adenitt og faryngitt er typisk. Periodisk feber kommer regelmessig hver 2-8 uke. Episodene varer 3-5 dager og omfatter after og sår i munn og svelg, ofte med hovne lymfeknuter på halsen. Hodepine og abdominale smerter. Veksten påvirkes ikke.
Blodprøver viser leukocytose med neutrofili, økt CRP, SR og fibrinogen. Enkelte har forhøyet IgD.
Genetikk: Polygenetisk til forskjell fra de andre autoinflammatoriske sykdommene som skyldes mutasjon i ett enkelt gen.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, Juvenil idiopatisk artritt (JIA), spesielt systemisk JIA, Kawasaki sykdom, tilbakevendende virusinfeksjoner, bakterielle infeksjoner-
Behandling: Spesielt effektiv behandling har en ikke. Behandling med kortikosteroider i korte perioder kan være aktuelt, men risiko for bivirkninger begrenser bruken. Kolkisin har effekt hos noen (Quintana-Ortega C, 2020). H2-blokker i form av cimetidin skal være effektiv hos ca. 30% av pasientene (Feder H, 1992). Il-1 hemmer (anakinra) kan utprøves. Også tonsillektomi kan vurderes.
Litteratur: Vanoni F, 2016
ROSAH syndrom (NF-κB-mediert)
ROSAH syndrom skyldes mutasjon i ALPK1-genet. Kliniske kjennetegn er retina-dystrofi, opticus-ødem, splenomegali og anhidrose, samt artritt, migrene og kalk i basalganglier (Kozckli, 2022).
Schnitzlers syndrom
Schnitzlers syndrom kjennetegnes ved urtikarielt, kløende utslett og økt mengde IgM-κ (sjelden IgG) paraprotein i serum (Simon A, 2013). Tilstanden er omtalt i et eget kapittel.
Debutalder: Voksne, median 60 år.
Spondyloartritt
Denne sykdomsgruppen spondyloartritt som omfatter Bekhterevs sykdom (ankyloserende spondylitt), psoriasisartritt, reaktiv artritt og IBD-relatert artritt har patogenetisk vist seg å ha både autoimmune og autoinflammatoriske trekk (Sibley CH, 2016; Bilgin E, 2022).
Debutalder: Barne-voksen alder, sjelden debut etter 50 års alder
Stills sykdom i voksen alder (adult Stills)
Sykdommen har innslag av autoinflammasjon. Episoder med feber og høy inflammasjon. I patogenesen ses en pro-inflammatorisk kaskade der det innate immunsystemet er aktivert, men til en viss grad også det adaptive immunsystemet (Feist E, 2018). Signalmolekylene (interleukiner) IL-1β, IL-6, IL-17, TNF og IL18 er forhøyet (Pascal V, 2005 ; Kudela D, 2019). Polygenetisk genese. Behandling med IL-1 hemmere (anakira, canakinumab) har ofte god effekt. Vennligst les mer om Stills sykdom i voksen alder i eget kapittel.
Debutalder: Oftest unge voksne, selv om sjeldne tilfeller i høy alder er beskrevet
TNF reseptor assosiert periodisk syndrom (TRAPS) (Hibernian fever)
ICD-10 M04.8 (andre autoinflammatoriske sykdommer)
Sykdomsårsak: Autosomalt dominant arvelig sykdom som angriper barn. Tilstanden ble tidligere kalt Familial Hebernian Fever og skyldes mutasjon i TNFR1 (TNF reseptor-1) genet (kromosom 12). Tilstanden medfører økte nivåer av proinflammatoriske cytokiner inkludert IL-1β, TNFα og IL-6.
Prevalens: 1 pr million
Debutalder: Sykdomsstart i tidlig barnealder er vanligst med median debutalder 4,3 år. Imidlertid diagnostiseres ca. 10% av tilfellene hos personer over 30 års alder, da vanligvis med mildere varianter av sykdommen.
Symptomer: TRAPS gir tilbakevendende feber, migrerende erythem (etterlater ofte ekkymoser), abdominal smerte (peritonitt, diare eller obstipasjon), artritt (store ledd), konjunktivitt, uveitt, periorbitalt ødem, myalgi, pleuritt-smerter, artralgi, evt. migrerende ødem pga. fasciitt og hodepine, oppkast og diare. Periodene kommer hver 4-6 uke og varer 4-21 dager. Noe økt risiko på sikt for amyloidose.
Genetikk. Diagnosen kan sikres ved gentest av TNFRSF1A genotype.
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, juvenil idiopatisk artritt (JIA), spesielt systemisk JIA, inflammatorisk tarmsykdom (IBD), juvenil systemisk lupus erythematosus (jSLE), tilbakevendende virusinfeksjoner, osteomyelitt.
Behandling: Biologiske legemidler er aktuelle. TNF-hemmer (etanercept), IL-1 hemmer (anakinra, canakinumab) eller IL-6 hemmer (tocilizumab) har ofte effekt slik at sykdomsaktiviteten faller, risiko for amyloidose og progressiv organskade synker (Cudrici C, 2020).
Urinsyregikt
En kan definere urinsyregikt som en ervervet autoinflammatorisk sykdom. Autoinflammasjonen skyldes inflammasom aktivitet som utløses av urinsyrekrystaller. Anfall kommer anfallsvis, kan være ledsaget av feber og utløst av traume. Menn angripes hyppigere enn kvinner.
Debutalder: Oftest blant voksne. Ved sjeldne genetiske varianter kan sykdommen utløses hos barn.
Behandling med blant annet Kolkisin og IL-hemmere (anakinra, canakinumab). Vennligst les mer om i urinsyregikt i eget kapittel.
VEXAS syndrom
Sykdommen er en autoinflammatorisk sykdom, men ikke et periodisk febersyndrom. VEXAS skyldes ervervet gen-mutasjon i somatiske celler (ikke arvelig) i voksen alder og preges av vedvarende inflammasjon hos eldre menn. Vennligst les om VEXAS i eget kapittel.
Yao syndrom (YAOS)
Symptomer: YAOS ble først beskrevet i 2011 og kjennetegnes ved periodisk feber, dermittt, poilyartritt, gastrointestinale symptomer og sicca med ødematrøse øyelokk.
Årsak. Syndromet er relatert til spesifikke varianter av NOD2. Nucleotide-binding oligomerization domain-containing protein-2 (NOD2) er en NOD-lignende reseptor (NLR) i cytosol som ble påvist i 2001. Mutasjoner i NOD2 genet assosieres med Crohns sykdom, Blau syndrome og Yao syndrome (YAOS)/ NOD2-associated autoinflammatory disease (NAID)
Differensialdiagnoser: Andre autoinflammatoriske sykdommer/periodiske febersyndromer, inflammatorisk tarmsykdom (IBD), sarkoidose, Juvenil idiopatisk artritt (JIA), juvenil systemisk lupus erythematosus (jSLE), tilbakevendende virusinfeksjoner.
Debutalder: Voksne
Litteratur: Yao Q, 2021
Retningslinjer
EULAR/ACR: Romano M, 2022 (diagnose og behandling av IL-1 relaterte tilstander)
Litteratur
- Krainer J, 2020
- Szekanecz Z, 2021
- De Jesus AA, 2013
- Grateau G. Rheumatology 2004
- Yao & Furst. Rheumatology 2008
- Touiton. Best Pract Res Clin Rheum 2008
- Grateau & Durum. Best Pract Res Clin Rheum 2010
- Henderson & Goldbach-Mansky. Curr Opin Rheum 2010
