ANDRE SYKDOMMER (REV 063-REV 077)
153 Fabrys sykdom (REV 073)
Øyvind Palm and Jan Tore Gran
Kjennetegn på Fabrys sykdom
Fabrys sykdom kan debutere med diverse uspesifikke symptomer som ofte begynner i tidlig barndom, men diagnostiseres noen ganger først blant voksne.
Uklare smerte i armer, ben og i mage/tarm
Unge voksne med CNS manifestasjon assosiert med myokardinfarkt og redusert nyrefunksjon
Genetisk sykdom med mangel på enzymet alfa-galaktosiderase (alfa-Gal A)
Gutter er sterkere angrepet enn kvinner
Lysosomale lagringssykdommer er ellers omtalt i eget kapittel.
Metabolske tilstander og hemokromatose er omtalt i gene kapitler. Skjelettsykdommer er beskrevet i ulike kapiteler, for eksempel under skjelett-tumorer, osteonekrose og osteopetrose eller osteomyelitt.
ICD-10: E75.2
Definisjon
. CC BY 2.0")
Fabrys sykdom er den vanligste arvelige lysosomale lagringssykdommen.
Den karakteriseres ved mangel på enzymet alfa-galaktosidase A (α-Gal A). Dette fører til opphopning av globotriaosylceramid (Gb3) i kroppens celler, noe som forårsaker skade i ulike organer. Sykdommen er X-bundet recessiv, hvilket betyr at kvinner er bærere av sykdommen, mens menn oftest rammes. Symptomene er ofte diffuse og kan ligne på revmatiske sykdommer, derav kallenavnet «den store imitator» (Bokhari SRA, 2023).
Tilstanden håndteres av ulike spesialister som pediater, endokrinolog, hjerte- nyre- øye- og hud-leger. Revmatologens rolle er å skille tilstanden fra revmatologiske diagnoser.
Sykdomsårsak
Fabrys sykdom skyldes genetisk mutasjon på X-kromosomet som fører til mangel på α-Gal A, noe som medfører akkumulering av glykosfingolipider (globotriaosylceramide, Gb3) i nerver, hud, nyrer, hjerte, hjerne, øyne og skjelett. Det er funnet hundrevis av mutasjoner i genet for α-Gal A som alle kan forårsake Fabry sykdom og er lokalisert på X-kromosomet (Bokhari SRA, 2023).
Forekomst
Fabrys sykdom forekommer i alle aldersgrupper og etnisiteter. Menn angripes (X-chromosomal recessiv arv). Den estimerte kliniske prevalensen varierer mellom studier fra 1/17.000 til 1/117.000. I Norge er det kjent rundt 100 tilfeller.
Symptomer
Fabrys sykdom kan gi et bredt spekter av symptomer som ofte starter i barndommen. Tidlige tegn inkluderer nevropatiske smerter i hender og føtter, gastrointestinale smerter, samt hudforandringer. Sykdommen kan også affisere en rekke andre organer (Ortiz A, 2018):
- Abdominalt. Smerter på grunn av mesenterial iskemi eller nevrogene smerter.
- Hjerte: Rytmeforstyrrelser. Hjerteklaffskader (aorta- og mitralklaffer)
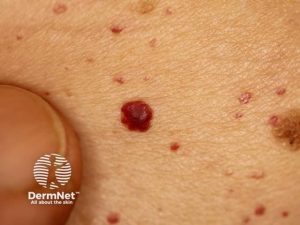
- Hud: Angiokeratomer er små, ikke smertefulle kuler (papler/noduli), oftest på lår, magen (ved umbilicus) og over bekkenområdet. Teleangiektasier. Anhidrose (mangel på svette). Redusert spytt- og tåre produksjon: Differensialdiagnose Sjøgrens syndrom, Sarkoidose. Raynauds-lignende hvite og kalde hender eller brennende smerter (som er utypisk for Raynauds fenomen).
- Nevropati: Begynner gjennomsnittlig ved 10 års alder: Brennende smerter, særlig i hender og føtter (tynnfibernevropati). Episoder med sterk smerte/smertekrise. Tinnitus (øresus). Svimmelhet. Hjerneslag i relativt ung alder. Differensialdiagnose: DaDA2.
- Nyrer: Protein i urinen (som kan skumme). Differensialdiagnose: Autoimmun nyrebetennelse ved blant annet systemisk lupus (SLE) og JSLE, nyrecyster. Progredierende redusert nyrefunksjon.
- Luftveier/lunger: Dyspne, stridor, tørrhoste, snorking.
- Skjelett: Osteoporose: Differensialdiagnose: Andre årsaker til osteoporose blant barn. Bennekroser
. CC BY 2.0")
- Øyne: Cornea verticillata (Hornhinnene blir uklare): Kan være debutsymptom. Retinal vaskulopati, katarakt, sentral retinal arterie okklusjon (sjelden), redusert tåre sekresjon.
- Annet: Redusert hørsel, Utmattelse (Fatigue), abdominale smerter, vekttap.
Undersøkelsesfunn
Anamnese. Familieanamnese er aktuell på grunn av den X-kromosomale arveligheten. Redusert svette- og tåreproduksjon forekommer. Polydipsi og polyuri. En historie med alvorlig hjertemanifestasjoner, CNS-symptomer, svimmelhet, smertefulle pareser, abdominale kramper, diare, påvirket mikrosirkulasjon i huden og uklart syn og redusert hørsel.
Klinisk undersøkelse kan avdekke hypertoni med nedsatt nyrefunksjon, redusert tåreproduksjon (Schirmers test), kardiomyopati, rytmeforstyrrelser, smertefulle pareser i ekstremiteter, petekkier i huden, uklar linse og corneaforandringer i øyne. Angiokeratomer (mellom navle og lår), hyperhidrose, proteinuri, hjerteaffeksjon, affeksjon av sentralnervesystemet, diare, evt. parestesier, osteoporose, osteonekrose og Raynaud-fenomener.
Laboratorieprøver kan omfatte elektrolytter og nyrefunksjonsprøver. Urin-sediment for fettlegemer. Spesifikk diagnose kan gjøres dersom det ved enzym analyse påvises for lav alpha-Gal A aktivitet i lymfocytter eller plasma.
Gen-analyser i EDTA blod er mulig, men ikke alltid konklusive. Kan sendes Avdeling for medisinsk genetikk, OUS Ullevål.

Bildediagnostikk består av røntgen thoraks, CT, CT-angiografi, MR, MR-angiografi og M-spektroskopi.
Andre. EKG og ekkokardiografi for kartlegging av hjertemanifestasjoner som rytmeforstyrrelse og patologiske strukturelle forandringer.
Biopsi fra hud eller nyrer kan bidra til diagnosen. Depoter av glykolipider er typisk. Elektronmikroskopi av nyrevev viser concentriske lag med inklusjoner i form av “zebra legemer”.
Diagnosen
Diagnosen stilles basert på sykehistorie, kliniske funn og laboratorieundersøkelser. Måling av α-Gal A-aktivitet er avgjørende for å bekrefte diagnosen. Genetisk testing kan også være nyttig.
Differensialdiagnoser
På grunn av det brede spekteret av symptomer er det viktig å vurdere en rekke differensialdiagnoser (Böttcher T, 2013; Hiltz MJ, 2018), inkludert:
- DADA2 (juvenil PAN): Sjelden autoinflammatorisk vaskulitt som kan gi hudmanifestasjoner, nevrologiske symptomer og feber. Kan forveksles med Fabrys sykdom på grunn av hudmanifestasjoner og nevrologiske symptomer.
- Dermatomyositt hos barn (JDM): Angriper muskler og hud, og kan gi utslett, muskelsvakhet og leddsmerter. Kan forveksles med Fabrys sykdom på grunn av hudmanifestasjoner og leddsmerter.
- Erythromelalgi: Sjelden nevrologisk lidelse som gir brennende smerter og rødhet i ekstremiteter. Kan forveksles med Fabrys sykdom på grunn av brennende smerter og rødhet i ekstremiteter.
- Fibromyalgi: Kronisk smertetilstand som gir utbredte smerter, tretthet og ømhet i kroppen. Kan forveksles med Fabrys sykdom på grunn av kroniske smerter og tretthet.
- Irritabel tarmsyndrom: Funksjonell tarmsykdom som gir magesmerter, oppblåsthet og endret avføringsmønster. Kan forveksles med Fabrys sykdom dersom pasienten har gastrointestinale symptomer.
- Juvenil artritt (JIA): Fellesbetegnelse for flere former for barneleddgikt, som kan gi leddsmerter, hevelse og stivhet.
- Hypertrofisk kardiopati av andre årsaker: Hjertesykdom som gir fortykkelse av hjertemuskelen, og kan gi symptomer som brystsmerter og tungpust.
- Lakunære syndromer: Små, subkortikale cerebrale infarkter i hjernen som kan gi ulike nevrologiske symptomer. Forårsaket av arterieokklusjon. Kan forveksles med Fabrys sykdom dersom pasienten har nevrologiske symptomer.
- Menieres sykdom: Sykdom i det indre øret som gir anfall med svimmelhet, hørselstap og tinnitus. Kan forveksles med Fabrys sykdom dersom pasienten har vestibulære symptomer.
- Multiple sklerose (MS): Autoimmun sykdom som angriper sentralnervesystemet, og kan gi en rekke nevrologiske symptomer. Kan forveksles med Fabrys sykdom dersom pasienten har nevrologiske symptomer.
- Nyresykdom av andre årsaker: Sykdom i nyrene som kan gi symptomer som tretthet, hevelser og endret vannlating. Kan forveksles med Fabrys sykdom dersom pasienten har nyreproblemer.
- Nevropati av andre årsaker: Skade på nerver i armer og ben, som kan gi smerter, nummenhet og prikking. Kan forveksles med Fabrys sykdom dersom pasienten har nevropatiske smerter.
- Revmatisk feber: Artritt som kan oppstå etter en streptokokkinfeksjon, og som kan gi leddbetennelse, hudutslett og hjerteproblemer. Kan forveksles med Fabrys sykdom dersom pasienten har ledd- og hudproblemer.
- Sinusvenetrombose: Trombose i en av de store venene i hjernen, som kan gi hodepine, kvalme og nevrologiske utfall. Kan forveksles med Fabrys sykdom dersom pasienten har nevrologiske symptomer.
- Slag: Oppstår når blodtilførselen til hjernen blir forstyrret. Kan forveksles med Fabrys sykdom dersom pasienten har akutte nevrologiske utfall.
- Teleangiektasier av andre årsaker: Utvidelse av små blodkar i huden, som kan gi røde eller blålige flekker. Kan forveksles med Fabrys sykdom dersom pasienten har teleangiektasier.
Behandling
Behandlingen av Fabrys sykdom kan omfatte tre aspekter: (a) enzym substitusjon for å kontrollere sykdomsprogresjon. (b) Supplerende smertebehandling med analgetika mot kronisk nevropati og akutte nociceptive smerter, inflammasjon eller en blanding av disse. (c) Modifisert / tilpasset livsstil (Politai JM, 2016).
Enzym-terapi (alfa-galaktosidase-A) kan stanse sykdomsutviklingen og delvis reversere symptomer. Genterapi er under utprøving (Palaiodinou L, 2023). Symptombehandling. Medikamenter mot smerte eller kramper. Behandle nyresvikt .Pacemaker eller ICD (defibrillator) ved enkelte hjerterytmeforstyrrelser. Økt risiko for slag gjør at mange får profylakse med acetylsalisylsyre (ASA) eller annen antikoagulasjon.
Prognose
Dersom pasienten har hatt slag, foreligger risiko for flere slag i forløpet. Nyresvikt med transplantasjon kan bli nødvendig. Terminal hjertesvikt og leversvikt forårsaker prematur død hos noen. Kvinner som er heterozygote har en mye mildere sykdom sammenlignet med menn.
Litteratur
Palaiodinou L, 2023 (behandling)
