Læringsmål REV 022. Revmatologen skal ha god kunnskap om kliniske bilder, herunder atypiske kliniske bilder, samt differensialdiagnostikk/diagnoser ved mistenkt systemisk bindevevssykdom.
Læringsmål REV 038. Differensialdiagnoser ved vaskulitt. Revmatologen skal ha god kunnskap om differensialdiagnostikk/diagnoser ved samtidige symptomer fra multiple organer/vev ved systemiske vaskulittsykdommer.
Transplanterte organer (benmarg, nyrer, hjerte, andre) fra en annen person (allogent transplantat) kan aktivere immunsystemet slik at sykdom oppstår (Aladag E, 2020). Tilsvarende kan også skje etter blodtransfusjoner eller etter kontakt med andre fremmede stoffer, inklusiv medikamenter.
Graft-versus-host disease (GVHD) er en systemisk sykdom som oppstår når graftet (vevet som blir transplantert) blir gjenkjent som fremmet og angrepet av mottakerens egne immunceller (Socie G, 2014). Transplantasjon av eget vev (autologt transplantat) eller vev fra enegget (genetisk identisk) tvilling utløser derimot ikke GVHD. GVHD medfører symptomer, hyppigst fra hud, øyne, munnhule, mage-tarm, genitalia, lunger, muskler, fascier og ledd (Rørvik SD, 2023).
Symptomene kan feiltolkes som revmatiske bindevevssykdommer, særlig systemisk lupus eller kutan vaskulitt. GVHD utredes og behandles ofte av immunologer eller transplantasjonsmedisinere. Tilstanden defineres som en akutt avstøtnings-reaksjon når den oppstår innen 100 dager (Aladag E, 2020), senere som kronisk GVHD (Hamilton GK, 2021. Overlappende tilstander mellom disse forekommer også. Sykdommen håndteres av hematolog, immunolog og spesialister på de de angrepne organer. Revmatologens oppgave er å skille tilstanden fra revmatiske sykdommer.
Patogenese
GVHD oppstår når organmottakerens immunsystem reagerer på uforlikelighet. Dette kan skje etter en allogen transplantasjon (mellom individer av samme art), selv om en forebygger med immunsupprimerende medikamenter. Risikofaktorer er vevstypeuforlikelighet mellom donor og pasient, bruk av mobiliserte stamceller fra perifert blod, tidligere akutt GVHD, høy pasientalder, kvinnelig donor til mannlig pasient og mangelfull profylakse som reduserer andelen T-celler (Atkonson K, 1990). Profylakse med antitymocytt-immunglobulin reduserer risikoen for GVHD (Rørvik SD, 2023). Den immunologiske reaksjonen er ikke helt forstått, men donors T-celler, antigen-presenterende celler (APC) og B-celler av betydning der også cytokiner, kemokiner og andre signalsubstanser bidrar (Magenau J, 2016). Kronisk GVHD medfører vedvarende inflammasjon og fibrose i ulike organer.
Epidemiologi
Forekomsten er størst etter allogen benmargstransplantasjon (ved behandling av systemisk sklerose med HMAS benyttes autolog transplantasjon). Til tross for profylakse og HLA-forlikelighet ses akutt GVDH hos 30-50% etter allogen hematopoetisk stamcelle transplantasjon (benmargstransplantasjon med ikke-eget vev) fra første grads slektning (Al-Kadhimi Z, 2014). Forekomsten er høyere når vev stammer fra ikke-matchede donorer. Mange av tilfellene forløper kronisk. Insidens av kronisk GVHD varierer mellom 6% og 80% (Atkinson K, 1990). Data fra Oslo Universitetssykehus viste at forekomsten sank fra 47% til 19% etter innføring av profylakse med antithymocytt-immunglobulin (Vo DD, 2023; Jacobsohn DA, 2012).
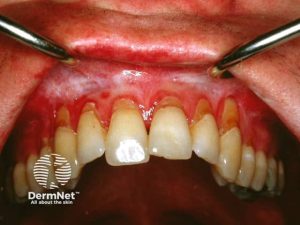
Oral GVHD med røde og hvite flekker, noe som indikerer betennelse eller forandringer i vevet. Illustrasjon: Dermnet. CC BY-NC-ND 3.0 NZ
Symptomer
Symptomer oppstår oftest 3-12 måneder etter transplantasjon. Hos ca. 10% debuterer manifestasjonene senere. Manifestasjoner fra mage og tarm, hud og feber er vanligst. Vanligvis angripes ett organ, selv om kombinasjoner forekommer (Ferrara JL, 2009) (Vaillant AAJ, 2024).
Hud-symptomer foreligger hos 67%.Makulopapulært eksem på hals, ører, skuldre håndflater og fotsåler. “Solbrent” hud. Blemmer og utslett over hele kroppen er mer alvorlige symptomer (se foto ovenfor). Ved kronisk GVHD kan huden bli indurert på grunn av fibrose, noe som kan minne om systemisk sklerose eller lokalisert skleroderma/morfea. Pigmenttap, hårtap og negledystrofi kan også ses.
Gastrointestinale symptomerses hos ca. 30%. Sår i øsofagus, diare, kvalme, oppkast, abdominale knipende smerter er vanlig.
Genitale sår (lichen planus-lignende, ulcerasjoner, arr-forandringer) ses hos 12%.
Infeksjonerer en komplikasjon som kan relateres til behovet for immunsuppressiv medikasjon.
Leverangripes hos ca. 44%. Leverenzymer i blodet stiger. ALP og konjugert bilirubin stiger først (ikterus).
Lunge-manifestasjoner ses hos 50%. Ny belastningsdyspne, tørrhoste kan være tegn på relatert bronchiolitis obliterans som er en karakteristisk manifestasjon.
Muskler og ledd angripes hos 29%. Myositt, fasciitt og sklerose med kontrakturer kan forekomme.
Orale sår ses ved kronisk GVHD hos ca. 60%. Munntørrhet og ulcerasjoner er vanlig. Kronisk forløp kan ligne lichen planus, men kan også utvikle seg til plateepitel karsinom.
Sicca-symptomer. Tørr munn og tørre øyne (Sjøgren syndrom-lignende) forekommer ved kronisk GVHD. Keratokonjunktivitis sicca er en indikator for dårlig prognose.
Øyne angripes hos 48%. Smerte, røde øyne og tørrhet er typisk.
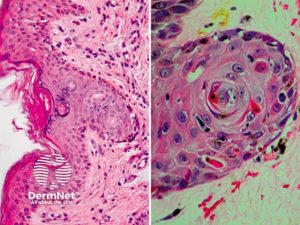
GVHD: Histologisk vevsundersøkelse forventer å vise: Økt tilstedeværelse av lymfocytter i de berørte vevene. Skade på epitelcellene som kler organene, som for eksempel huden, mage-tarmkanalen eller leveren. Tegn på inflammasjon, som rødhet, hevelse og økt blodtilførsel. Ved kronisk GVHD kan det være tegn på fibrose (arrdannelse) i de berørte vevene. Illustrasjon: Dermnet. CC BY-NC-ND 3.0 NZ
Differensialdiagnoser
GVHD kan etterligne en rekke andre tilstander, og det finnes ingen enkel, “gullstandard” test for å bekrefte GVHD. Derfor er det essensielt å vurdere og utelukke andre mulige årsaker til pasientens symptomer.
Feber av “ukjent årsak”:Feber uten en klar årsak kan være et symptom på GVHD, men kan også skyldes en rekke andre tilstander og krever ytterligere utredning for å skille fra GVHD.
Hud/eksem og slimhinnesår
Behcets sykdom: En sjelden inflammatorisk sykdom som kan forårsake tilbakevendende sår i munn og underliv, samt hudlesjoner, og som i likhet med GVHD kan gi hudutslett og sår, men som også har andre symptomer og lesjoner.
Dermatomyositt: Kan ligne på GVHD-syndrom fordi begge tilstandene kan involvere hudutslett, muskelsvakhet og systemisk betennelse.
Eksem utløst av medikamenter/ DRESS.: Utslett forårsaket av medikamenter kan ligne på GVHD-utslett, men oppstår som en reaksjon på et spesifikt medikament og vil vanligvis forsvinne når medikamentet seponeres.
Stråle dermatitt (kreftbehandling): Hudirritasjon forårsaket av strålebehandling kan ligne på GVHD-utslett, men oppstår i områder som er utsatt for stråling og har et annet tidsmessig forhold til behandlingen.
Virus-infeksjoner: Ulike virusinfeksjoner kan gi hudutslett som ligner på GVHD, men som ofte ledsages av andre symptomer som feber, hoste og sår hals og som har et annet forløp enn GVHD.
Infeksjoner med diare; nfeksjoner i mage-tarmkanalen kan forårsake diaré, som også er et vanlig symptom på GVHD, men som ofte ledsages av andre symptomer som feber, oppkast og magesmerter, og som har en annen årsak enn GVHD.; clostridium, CMV- eller Epstein barr virus reaktivering, adenovirus, rotavirus, mycobacterium avium kompleks, giardia, cryptosporidium.
Trombotisk mikroangiopati: En tilstand som kjennetegnes av dannelse av små blodpropper, som kan påvirke mage-tarmkanalen og gi symptomer som diaré og magesmerter, og som i likhet med GVHD kan gi symptomer fra mage-tarmkanalen, men som har en annen patofysiologi.
Gallesalet malabsorpsjon: En tilstand der kroppen har problemer med å absorbere gallesalter, noe som kan føre til diaré, og som i likhet med GVHD kan gi diaré, men som har en annen årsak og mekanisme.
Kvalme, oppkast, anoreksi: Kvalme, oppkast og tap av appetitt kan være bivirkninger av ulike behandlinger som kjemoterapi, immunterapi, stråling, antibiotika eller opioider og kan ligne på symptomer forårsaket av GVHD i mage-tarmkanalen.
Viral hepatitt (Hepatitt B, Hepatitt C). Infeksjon med hepatittvirus kan forårsake leverbetennelse og lignende symptomer som ved GVHD i leveren, men som har en annen årsak og forløp.
Medikamentindusert toksisitet og DRESS syndrom: Skade på leveren forårsaket av medikamenter kan ligne på GVHD i leveren, men er en reaksjon på et spesifikt medikament, og vil vanligvis forsvinne når medikamentet seponeres.
Immunterapi-indusert hepatotoksisitet (ved kreft-behandling): Noen former for immunterapi kan skade leveren og gi lignende symptomer som ved GVHD i leveren, men er relatert til immunterapibehandlingen.
Sinusoidalt obstruktivt syndrom (okklusjon av små levervener): En tilstand som involverer blokkering av små blodårer i leveren, og som kan gi lignende symptomer som ved GVHD i leveren, men som har en annen mekanisme.
Sjokk-lever: En tilstand med akutt leverskade som kan skyldes ulike årsaker som infeksjon eller hjertesvikt, og som kan gi lignende symptomer som ved GVHD i leveren, men som har en annen årsak.
Sjøgrens syndrom (sicca-fenomen): Kan gi tørre øyne og tørr munn, og som i likhet med GVHD kan gi tørrhet i slimhinner, men som har et annet mønster av symptomer.
Systemisk lupus erythematosus (SLE): Kan påvirke flere organer og gi ulike symptomer, hvorav noen kan ligne på GVHD, men som har også andre symptomer og en annen patofysiologi.
Behandling
Behandlingen tar sikte på å redusere symptomer og organskade. Spesielt er utvikling av lungesvikt viktig å forhindre. På lengre sikt kan immunologisk toleranse utvikles, slik at behandlingen kan trappes ned. Ved mild sykdom kan lokalbehandling være tilstrekkelig.
Kortikosteroider. Systemisk behandling med kortikosteroider (initialt 0,5-1mg/kg) er første valg ved akutt GVHD (Goker H, 2001). Imidlertid responderer 35-50% av pasientene ikke tilstrekkelig og trenger supplerende tiltak (Murata M 2015). Valg av annen-linje behandling baseres på effekt tidligere behandling og potensiell toksisitet (Martin PJ, 2012).
T-celledeplesjon: I noen tilfeller fjernes T-celler fra den transplanterte stammen for å redusere risikoen for GVHD (Bleakly M, 2022).
Infeksjonsprofylakse med valaciklovir mot virus og sulfametoksazol og trimetoprim mot pneumocystis jirovecii anbefales. Ved høye doser prednisolon (>20 mg/d kombinert med annen immunsuppressiv behandling), benyttes også posakonazol mot muggsopp (Rørvik SD, 2023).
Prognose
Ved kronisk GVHD og behov for systemisk behandling er median behandlingsvarighet ved steroid-resistens 2-3 år. Omtrent 50% kunne avslutte behandlingen innen syv år (Vigorito AC, 2009; Rørvik SD, 2023).).
Kliniske risikofaktorer for langvarig tilstand er uttalte hud-manifestasjoner, diare, trombocytopeni, økte leverenzymer, lunge- eller lever-manifestasjoner (Müller JA, 2016). Risikoen for GVHD er høyere etter transplantat fra donorer som ikke er blant HLA-identisk søsken eller anen nær slektning. Andre risikofaktorer er høy alder blant pasient eller donor, kvinnelig donor, perifert blod som stamcelle kilde (Zeiser R, 2017), tidligere GVHD og seropositivitet for cytomegalovirus og Epstein Barr virus. Omtrent 14-36% utvikler alvorlig akutt GVHD (Al-Kadhimi Z, 2014).
Lunge-komplikasjoner over tid er bronchiolitis obliterans syndrom, interstitiell lung sykdom (ILD), obliterativ bronchiolitt, organiserende pneumoni og pleuroparenchymal fibroelastosis (Bergeron A, 2017)
Gastroenterologisk ses fibrose, motilitetsforstyrrelser og malabsorpsjon.
Lever-komplikasjoner omfatter endotelcelle-inflammasjon, gallegang-destruksjon og pericholangitt (Ferrara JL, Lancet 2009).