ANDRE SYKDOMMER (REV 063-REV 077)
167 IgG4 Relatert sykdom (multifokal fibrosklerose) (REV 064)
Øyvind Palm and Jan Tore Gran
Kjennetegn på IgG4 relatert sykdom
Sklerotisk benign tumor som kan fortrenge, komprimere og skade tilgrensende organer.
Lymfeknuter, spytt- og tårekjertler, pankreas og retroperitonealt vev angripes hyppigst, men alle lokalisasjoner er mulig.
God allmenntilstand.
IgG4 i serum oftest forhøyet i aktiv fase, men er ikke spesifikk eller sensitiv. Sikker diagnose ved biopsi.
God effekt av kortikosteroider, selv i lave doser.
ICD-10: M35.5 Multifokal fibrosklerose
ATC koder (for legemiddelstatistikk): Prednisolon: H02A B06 Immunsuppressive legemidler: L04A A
Definisjon
IgG4-relatert sykdom (IgG4-RD) er en kronisk inflammatorisk tilstand karakterisert ved (Wallace ZS, 2024; Kamisawa T, 2014):
- Langsomt voksende, godartede fibro-inflammatoriske svulster i ett eller flere organer, inkludert tåre- og spyttkjertler, øyehulen, bukspyttkjertelen, galleveier, retroperitoneum, aorta, lymfeknuter og skjoldbruskkjertelen.
- Forhøyet nivå av IgG4 i blodet.
- God respons på behandling med glukokortikoider (steroider).
- Fravær av feber, artritt og forstørret milt.
Revmatologer spiller en viktig rolle i diagnostisering og behandling av IgG4-RD, spesielt når sykdommen rammer flere organsystemer. Samarbeid med andre spesialister er ofte nødvendig for å vurdere og behandle spesifikke organmanifestasjoner. Målet med konsultasjonen er å:
- Bekrefte eller avkrefte diagnosen IgG4-RD.
- Vurdere omfanget av sykdommen og hvilke organer som er affisert.
- Starte behandling med glukokortikoider og eventuelt andre immunsuppressive legemidler.
- Utarbeide en plan for oppfølging og monitorering av sykdomsaktivitet og behandlingseffekt.
Historie
Sykdommen vi i dag kjenner som IgG4-relatert sykdom (IgG4-RD), har en lang og interessant historie. Allerede i 1892 beskrev Johann von Mikulicz-Radecki en 42 år gammel bonde med symmetrisk hevelse i tårekjertlene, ørespyttkjertlene og kjertlene under kjeven, uten symptomer på tørrhet. Mikroskopisk undersøkelse av vevet viste massiv infiltrasjon av en type hvite blodceller kalt mononukleære celler (J. Mikulicz; “Über eine eigenartige symmetrische Erkrankung der Trähnen- und Mundspeicheldrüsen. Beiträge Zur Chir Festschr Gewidmet Theodor Billroth Stuttg Ferdinand Enke” (1892), pp. 610-630).
Det skulle gå over hundre år før neste milepæl i forståelsen av IgG4-RD. I 2001 ble det beskrevet en sammenheng mellom autoimmune betennelser i bukspyttkjertelen og høye nivåer av IgG4 i blodet (Hamano H, 2001). Dette ledet til at IgG4-RD ble anerkjent som en egen sykdom i 2003 (Kamisawa T, 2003).
I årene etter 2003 ble det stadig tydeligere at IgG4-RD kunne ramme nærmest alle organer i kroppen. Sykdommen ble hyppigst sett i de store spyttkjertlene, øyehulen, bukspyttkjertelen, galleveiene, lungene, hovedpulsåren, området bak bukhinnen, hjernehinnene, skjoldbruskkjertelen og lymfeknuter.
Før man forsto at dette var ulike manifestasjoner av samme sykdom, ble de oppfattet som separate tilstander. Mikulicz syndrom (symmetrisk sykdom i tåre- og spyttkjertler), Küttners tumor (betennelse i kjertler under kjeven), Riedels tyreoiditt (betennelse i skjoldbruskkjertelen), Ormonds sykdom (retroperitoneal fibrose) og autoimmun pankreatitt var alle diagnoser som senere ble samlet under paraplyen IgG4-RD.
Patogenese
Etiologien/årsaken til IgG4-relatert sykdom (IgG4-RD) er fortsatt ukjent, men man antar at det er en autoimmun sykdom der immunsystemet feilaktig angriper kroppens eget vev.
Selv om sykdommen kjennetegnes av høye nivåer av IgG4-antistoffer i blodet, antas ikke disse å være den direkte årsaken til sykdommen. Dette støttes av at IgG4-antistoffer ikke aktiverer komplementsystemet, ikke danner immunkomplekser, og at andre tilstander med høye IgG4-nivåer, som IgG4 myelom, ikke gir symptomer på IgG4-RD (Chen LYC, 2019).
I stedet ser det ut til at CD4+ T-celler, en type hvite blodceller, spiller en sentral rolle i sykdomsutviklingen. Disse cellene aktiverer andre immunceller, inkludert B-celler som produserer IgG4-antistoffer, og stimulerer dannelsen av bindevev (fibrose). Denne prosessen fører til at store mengder arrvev dannes og infiltrerer ulike organer, noe som kan fortrenge normalt vev og redusere organfunksjonen (Mattoo H, 2016).
Det er fortsatt mye forskning som gjenstår for å avdekke de nøyaktige mekanismene bak IgG4-RD. En bedre forståelse av sykdommens patogenese vil kunne bidra til utvikling av mer målrettede og effektive behandlingsmetoder.
På bakgrunn av sykdommens lymfocyttinfiltrater, fibrosemønsteret, aortitt og oblitererende flebitt klassifiserer noen IgG4-RD blant vaskulitt-sykdommene (Stone JH, EULAR, 2024).
Epidemiologi
Prevalens og insidens
Den nøyaktige forekomsten av IgG4-relatert sykdom (IgG4-RD) i befolkningen er ikke fullstendig kartlagt. Estimater antyder en prevalens på 5,3 per 100 000 og en insidens på 0,78-1,39 per 100 000 personår (Wallace ZS, 2023). Det er imidlertid grunn til å tro at sykdommen er underdiagnostisert, og at den reelle forekomsten kan være høyere (Chen LYC, 2019).
Alder og kjønnsfordeling
Alder og kjønnsfordeling varierer avhengig av hvilke organer som er rammet. Generelt er menn omtrent dobbelt så ofte rammet som kvinner. Hyppigst forekomst sees hos menn mellom 50 og 70 år, hvor autoimmun pankreatitt type 1 er den vanligste manifestasjonen. IgG4-RD er sjelden hos barn, men øyeaffeksjon (retroorbitalt eller tårekjertelen) og pankreasaffeksjon er beskrevet (Wallace ZS, 2023).

Symptomer
Sykdomsbildet ved IgG4-relatert sykdom (IgG4-RD) varierer avhengig av hvilke organer som er rammet. Symptomene kan være subtile og utvikle seg gradvis over tid, noe som kan gjøre diagnostiseringen utfordrende. (Wallace ZS, 2024).
Eksempler på symptomer ved affeksjon av ulike organer:
- Spyttkjertler: Tørr munn (sicca-syndrom).
- Øyehulen: Fremstående øyeeple (proptose) på grunn av orbital pseudotumor.
- Bihuler: Tap av luktesans (anosmi) ved betennelse i nese og bihuler.
- Arterier: Utposninger på blodårene (aneurismer) eller rift i arterieveggen (disseksjon).
- Nyrer: Kronisk obstruksjon av urinlederen (ureter) eller væskeansamling i nyrene (hydronefrose) på grunn av retroperitoneal fibrose. Nedsatt nyrefunksjon (tubulointerstitiell fibrose).
- Bukspyttkjertel: Nedsatt funksjon av bukspyttkjertelen (eksokrin og/eller endokrin pankreassvikt), som kan føre til problemer med fordøyelsen og regulering av blodsukkeret.
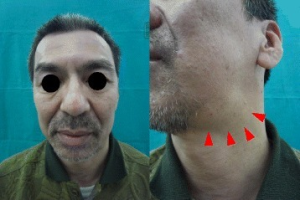
Ved affeksjon av overfladiske organer kan pasientene selv merke en eller flere harde, langsomt voksende svulster. Dette kan ramme områder rundt øynene (periorbitalt), spyttkjertelen parotis, submandibularis, lymfeknuter eller thyreoidea (skjoldbruskkjertelen).
Pasienter med IgG4-relatert autoimmun pankreatitt kan utvikle Ikterus, abdominale smerte og vekttap sent i forløpet. Også pankreasfunksjonssvikt (både eksokrin og endokrin) kan også forekomme. Feber, forstørret milt (over 14 cm) og artritt er uvanlig.
Respons på steroidbehandling. Et karakteristisk trekk ved IgG4-RD er den gode responsen på behandling med glukokortikoider (steroider) som prednisolon. Disse medikamentene demper betennelsen og fører ofte til reduksjon av svulstene. Sykdomsprogresjon under behandling med relativ høy dose prednisolon (30-40 mg/dag i 4 uker) er svært sjelden og kan tyde på en annen diagnose.
Kliniske kjennetegn i fire sentrale sykdomsmanifestasjoner av IgG4-RD (Wallace ZS, 2024)
| Pankreas og-hepatobilære sykdommer | Retroperitoneal og aorta | Hode- og hals- begrenset sykdom | Mikulicz og systemisk sykdom | |
|---|---|---|---|---|
| Typiske manifestasjoner | Autoimmun pankreatitt, skleroserende cholangitt | Retroperitoneal fibrose, aortitt, stor-kar-sykdom | Spytt- og tårekjertel forstørrelse, orbital manifestasjon | Klassisk symmetrisk tåre- og spyttkjertel-forstørrelse med manifestasjoner i thoraks og abdomen. |
| Mannlig predominans | Ja | Ja | Nei | Ja |
| Alder (gjennomsnitt, år) | 63 | 58 | 55 | 63 |
| Serum IgG4 konsentrasjon | Forhøyet | Normal til lett forhøyet | Forhøyet | Svært høy |
| Eksempler på feildiagnoser/lignende tilstander | Pankreascancer, autoimmun pankreatitt type 2, primær skleroserende cholangitt | Lymfom, Erdheim–Chester sykdom, temporalis arteritt | Sjøgrens sykdom og andre autoimmune bindevevssykdommer, granulomatose med polyangiitt/GPA, lymfom | |
Undersøkelser
IgG4-RD kan ramme et bredt spekter av organer og vev, noe som gjør diagnostiseringen utfordrende. Sykdommen kan etterligne kreft, infeksjoner og andre inflammatoriske tilstander. Tidlig diagnose er viktig for å unngå irreversible organskader.
Mistanke om IgG4-RD bør vekkes ved:
- Langsomt voksende svulster eller diffus hevelse i ett eller flere av de typiske organene.
- Uforklarlige symptomer som magesmerter, tørr munn, tørre øyne, gulsott eller nyreproblemer.
- Forhøyet IgG4-nivå i blodet.
- God respons på behandling med steroider.
Diagnosen bekreftes ved:
- Histologisk undersøkelse av biopsi fra affisert vev, som viser karakteristiske forandringer med IgG4-positive plasmaceller og fibrose.
- Bildediagnostikk (CT, MR, PET-CT) som kan visualisere svulster og organaffeksjon.
Utredningen initieres ofte av hevelser i parotis, lymfeknuter, tegn til autoimmun pankreatitt, eosinofili og hypergammaglobulinemi (Chen LYC, 2019). Vurder om typisk(e) organ er angrepet (pankreas, galleganger, orbita, tårekjertler, store spyttkjertler, retroperitoneum, lunger, nyrer, aorta, pachymeninger, thyreoidea /Riedels struma). Det er ikke typisk med feber, leukopeni, trombocytopeni eller manglende behandlingsrespons på kortikosteroider.
. CC BY-4.0")
Klinisk vurdering av overfladiske organer kan avdekke manifestasjoner i tårekjertler, orbita-nært, parotis, submandibularis, lymfeknuter på hals og andre lokalisasjoner, samt thyreoidea. Fibrøs, sklerotisk hevelse i eller omkring angrepne organer er typisk ved palpasjon. Allmenntilstanden påvirkes ved multiorgan-manifestasjoner. Vekttap ses særlig ved pankreasaffeksjon.
Laboratorieprøver. Rutinemessig kan det være aktuelt i utredningen å måle hemoglobin, leukocytter, trombocytter, CRP, nyre- lever- galle- pankreas- og thyreoidea-funksjonsprøver, Immunglobuliner med sub-klasser av IgG (inklusiv IgG4). Hormonprøver (TSH, veksthormon/somatotropin, anti-diuretisk hormon/ADH) ved mulig hypofyse-manifestasjoner. Urin-stiks. Fekal elastase 1 (FE-1) ved mulig pankreas-affeksjon.
Forhøyet serum IgG 4 > 1.4 g/L ses hos 70% – 80% av pasientene. Jo høyere IgG, desto større sjanse for at pasienten har IgG4-RD. IgG4-nivå på det dobbelte av øvre referanseområde (ofte benyttes referanseområde 0,03-2,010 g/L) er funnet å ha en spesifisitet på 99% for IgG4-relatert sykdom (Ghazale A, 2007). De høyeste verdiene ses ved Mikulicz syndrom (parotis og submandibularis) med systemisk manifestasjon. Multiorgan manifestasjon assosieres med høyere IgG4 i serum, men normale verdier utelukker ikke sykdommen. Lave verdier forventes ved pankreas-, lever/galle-manifestasjon eller retroperitoneal fibrose (Wallace ZS 2018). På den andre siden er forhøyet IgG4 et non-spesifikk funn. Hele 5% av befolkningen kan ha økt IgG4 i serum uten sykdommen. Høy IgG4/IgG ratio i serum. IgG4 utgjør > 10% av IgG totalt i serum. Normalt utgjør IgG4 mindre enn 5 % av alt IgG (referanse-verdi: 6,1-15,7g/L). Ved IgG4 sykdom ses ofte økte nivåer også av total IgG og IgE.
Perifer eosinofili er rapportert hos ca. 40%, da ofte kombinert med astma eller atopi. Forhøyet CRP og SR er også uspesifikke funn i likhet med positive anti-nukleære faktorer (ANA) og revmafaktorer (RF) hos noen. Serologisk forventes fravær av PR3 eller MPO-ANCA, SSA /Ro og andre spesifikke antistoff og kryoglobuliner. Lave komplementer kan relateres til immunkompleks-dannelse i nyrer og pankreas. Det er uvanlig med leukopeni, trombocytopeni og kryoglobuliner.
Høye verdier av plasmablaster (umodne plasmaceller) er også relatert til IgG4 sykdom. Målingene kan gjøres ved flow-cytometrisk analyse av B-celle populasjonen, men er ikke rutine. Andre diagnostiske metoder er ofte mer avgjørende i klinisk praksis. Referanseverdier fra friske kontroller: median 94, range 1-653/ml.
Bildediagnostikk. IgG4-relatert sykdom i angrepne organer kan visualiseres ved ultralyd, MR, CT eller PET/CT. Det er uvanlig med rask radiologisk progresjon, splenomegali eller patologiske funn i lange rørknokler (vurder differensialdiagnoser: histiocytose, Erdheim Chester). Ved pankreas-manifestasjon kan bildediagnostikk ikke skille sikkert mellom IgG4 relatert sykdom og malignitet, men typisk er at IgG4 relatert autoimmun pankreatitt ved MR viser en diffust forstørret pankreas med forsinket kontrastopptak og upåfallende avgrensning mot periferien. PET/CT er vist sensitivitet ved IgG4-relatert sykdom i arterier, spyttkjertler og lymfeknuter. Metoden kan dermed brukes til stadium-inndeling og monitorering (Ebbo M, 2014). Også den nyere FABI-PET/CT (via fibroblast aktiveringsprotein) kan vise fibrotisk aktivitet og dermed sykdomsaktivitet med fibrosedannelse ved IgG4-relatert sykdom (Mori Y, 2024). Endoskopisk retrograd cholangio-pankreatografi (ERCP) og endoskopisk ultralyd (EUS) kan være nyttige for biopsi-taking i pankreas og galleveier. I tillegg er ultralyd nyttig ved mistanke om nyremanifestasjon og behov for nyrebiopsi (Nabiar S, 2023).
. CC BY-2.0")
Biopsi er viktig for å sikre diagnosen (Deshpande V, 2012). IgG 4 relatert sykdom kjennetegnes ved tre klassiske funn: lymfoplasmacyttisk inflammasjon, fibrose med stråmatte/bølgelignende mønster og obliterativ venulitt. I tillegg kan keloid eller hyaline mønster ses. Granulomer er derimot uvanlig, slik at en da bør vurdere differensialdiagnoser (Nabiar S, 2021). IgG4 utgjør ved IgG4 sykdom >40% av totalt IgG, men ulikt i ulike organer. I enkelte tilfeller påvises ikke økt IgG4, muligens fordi sykdommen er i en mindre aktiv fase eller på grunn av behandlings-respons. “Stori-form” -fibrose (stråmatte-/bølgeligende). Lymfo-plasma-cellerike, tette celleinfiltrater med økt andel IgG4+ plasma celler uten granulomer eller nekrotiserende vaskulitt. Oblitererende flebitt (vene-okklusjon). Lett økt antall eosinofile leukocytter.
Tilstander som kan klassifiseres som IgG4 relaterte sykdommer
Ved en rekke tilstander sees assosiasjon til IgG4. Det skal imidlertid bemerkes at bare en viss andel av hver enkelt sykdomskategori er assosiert med IgG4. For eksempel er bare 50 % av alle idiopatiske abdominal-aortaaneurismer assosiert med IgG4 (Kasashima S, 2011). Av alle reseserte thorakal-aorta aneurismer vil bare 0,5-16 % være assosiert til IgG4.
- Abdominalt aortaaneurisme og andre aneurismer (Kasashima G, 2018)
. CC BY-NC 4.0")
Overlapping mellom de vanligste IgG4 relaterte sykdommene. Illustrasjon: Koizumi S, Kamisawa T, Kuruma S, Tabata T, Chiba K, Iwasaki S, Kuwata G, Fujiwara T, Fujiwara J, Arakawa T, Koizumi K, Momma K – Journal of Korean medical science (2015). CC BY-NC 4.0 - Aortitt. Definert ved at media-laget histologisk er angrepet (periaortitt når adventitia er involvert). Som ved periaortitt er infrarenale aorta oftest angrepet (Onkaramurthy NJ, 2023)
- Dakryoadenitt, kronisk skleroserende (ofte bilateral hevelse med lymfoid hyperplasi og fibrose) (Wang M, 2019)
- Eosinofil angiocentrisk fibrose (orbita og øvre luftveier) (Ahn J, 2018)
- Hypofysitt (autoimmun) (Decker L, 2016)
- Inflammatorisk pseudotumor i lunge (Zhu L, 2017)
- Kuttners tumor (tumor-hevelse av submandibularis) (Kaminski B, 2020)
- Skleroserende mediastinitt (Mediastinal fibrose) (Satochi T, 2020)
- Mesenteritt (skleroserende) (Lee SJ, 2016)
- Mikulicz sykdom (Kaminski B, 2020)
- Multifokal fibrosklerose = IgG4 relatert sykdom
- Pachymeningitt (Levraut M, 2019)
- Paranasal sinus fibrose (Bashyam M, 2018)
- Pankreatitt (autoimmun) type I (Basyal B, 2021)
- Ofte kombinert gallegangsstenose som ved skleroserende kolangitt
- Lymfadenopati hos 50%
- Ikterus, abdominale smerte og vekttap sent i forløpet
- Pankreas funksjonssvikt (eksokrin og endokrin) forekommer
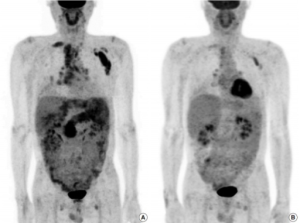
- Periaortitt definert ved at histologisk adventitia er angrepet med eller uten også media-manifestasjon.
- Oftest menn og da infrarenale aorta. Kvinner har oftere manifestasjoner over diafragma.
- Karakteriseres av et fibro-inflammatorisk vev som starter i de ytre lagene av abdominalaorta og iliaca-arteriene og som kan strekke seg inn i det omkringliggende vevet. Ofte uttalt affeksjon av adventitia, men ikke nødvendigvis media og intima. Derav betegnelsen periaortitt. Kan også affisere torakalaorta (Peng L, 2020)
- Perikarditt (konstriktiv) (Arao K, 2019)
- Retroperitoneal fibrose (Konno S, 2019)
- Riedels thyreoiditt (Dahlgren S, 2010)
- Subglottis stenose (idiopatisk) (Bach A, 2021)
- Sialoadenitt (kronisk skleroserende)
- Submandibularis eller parotis adenitt med
- Samtidig dakryoadenitt (Mikulicz sykdom)
- Spesielt ofte sees hevelse av submandibularis, polyklonal hypergammaglobulinemi og evt. hypokomplementemi. Ikke alle har sicca-symptomer, men ny tørrhet i munn- eller øyne kan være debut symptomer (Kaminski B, 2020)
- Skleroserende kolangitt (Tanaka A, 2019)
- Trakeal stenose (idiopatisk) (Gabrovska N, 2021)
- Tubulo-interstitiell nefritt (inflammatorisk pseudotumor i nyre) og TINU syndrom (Mann S, 2016; Joyce E, 2017)
Kriterier for diagnose
Japanske kliniske diagnostiske kriterier (sikker: 1 + 2 + 3. Sannsynlig: 1 + 3. Mulig: 1 + 2: (Nabiar S, 2021)).
-
Klinisk undersøkelse som viser typisk diffus/lokalisert hevelse eller masser i ett eller multiple organer.
-
Hematologisk undersøkelse viser forhøyet serum lgG4 konsentrasjon (over 135 mg/dL).
-
Histopatologisk undersøkelse viser: a) Markert lymfocytt og plasmacyttisk infiltrasjon og fibrose. b) Infiltrasjon av IgG4 + plasma celler: ratio avIgG4 +/ IgG + celler større enn 40% og større enn 10 IgG4 + plasma celler/HPF (high-power fields).
Klassifikasjonskriterier (ACR/EULAR konsensus kriterier)
Disse er omfattende, men er nyttige ved fordypning og studier (Wallace ZS, 2019). ACR/EULAR konsensus 2019: Wallace ZS, 2020 (spesifisitet 99,2-97,8%, sensitivitet 85,5-82,0%).
Differensialdiagnoser
- Autoimmune sykdommer
- Infeksjoner
- Kreft
- Primær skleroserende kolangitt forårsaker forhøyet IgG4 hos 10-15% (Boonstra K, 2014).
- -Autoimmun pankreatitt (non-IgG4 relatert): Kan forårsakes av en autoimmun reaksjon, men som ikke er assosiert med IgG4. Begge tilstandene kan gi hevelse i bukspyttkjertelen og forhøyede nivåer av visse immunglobuliner, noe som kan gjøre det vanskelig å skille dem fra hverandre.
- -Castlemans sykdom: En sjelden lymfoproliferativ tilstand som kan gi forstørrede lymfeknuter og systemiske symptomer. Begge tilstandene kan gi forstørrede lymfeknuter og systemiske symptomer, som feber og vekttap.
- -Crohns sykdom: Begge kan gi betennelse i mage-tarmkanalen, men IgG4-relatert sykdom rammer oftere andre organer i tillegg.
- –Eosinofili av andre årsaker, inkludert hypereosinofilt syndrom, allergier, infeksjoner og autoimmune sykdommer.
- –Kimura sykdom: kjennetegnes av forstørrede lymfeknuter i hodet og nakken, samt økte nivåer av eosinofile celler og IgE. Begge tilstandene kan gi forstørrede lymfeknuter og økte nivåer av eosinofile celler, men Kimura sykdom har vanligvis en mer karakteristisk lokalisering og andre funn, som økte IgE-nivåer.
- –Histiocytose (Rosai Dorfman, Erheim Chester): kjennetegnes av en økning i antall histiocytter, som er en type immuncelle. Begge tilstandene kan gi forstørrede lymfeknuter og systemiske symptomer, men histiocytose har en annen histopatologi og involverer andre immunceller.
- –Lymfadenopati av andre årsaker (sarkoidose, infeksjoner, autoimmune sykdommer og kreft)
- –Lymfom: kan gi forstørrede lymfeknuter og systemiske symptomer
- -Multicentrisk Castleman sykdom: Begge tilstandene kan gi forstørrede lymfeknuter og systemiske symptomer, men multicentrisk Castleman sykdom er ofte mer aggressiv og har en annen patogenese.
- –Orbital sykdom av annen årsak (GPA, EGPA/Churg-Strauss, Graves thyreoiditt, tumorer): Sykdom i øyehulen som kan skyldes ulike årsaker, inkludert autoimmune sykdommer og kreft. Begge tilstandene kan gi betennelse og hevelse i øyehulen, men den underliggende årsaken er forskjellig.
- –Plasma celle neoplasi (inklusiv IgG4 myelom): Kreft i plasmacellene, som produserer antistoffer. Begge tilstandene kan gi økte nivåer av IgG4, men plasmacelle neoplasi er en malign tilstand, mens IgG4-relatert sykdom vanligvis er benign.
- -Polyklonal hypergammaglobulinemi: Kan skyldes ulike årsaker. Begge tilstandene kan gi økte nivåer av immunglobuliner, men IgG4-relatert sykdom har vanligvis en mer spesifikk økning i IgG4-nivået.
- -Sarkoidose: Begge tilstandene kan gi forstørrede lymfeknuter og systemiske symptomer, men sarkoidose har en annen histopatologi og involverer andre organer oftere.
- -Sjøgrens sykdom: Kan gi forstørrede spyttkjertler, men Sjøgrens syndrom har andre karakteristiske funn, som tørre øyne og munn.
- –Ulcerøs kolitt: kan gi betennelse i mage-tarmkanalen, men ulcerøs kolitt er begrenset til tykktarmen og endetarmen, mens IgG4-relatert sykdom kan ramme andre organer i tillegg.
Behandling

Før behandlingen startes er det viktig at pasienten er oppklart om sykdommen, behandlingsmål og bivirkninger. Behandlingsmålet avklares i samråd med pasienten.
Behandling av IgG4-relatert sykdom avhenger av organmanifestasjonene og sykdommens alvorlighetsgrad. Behandlingsalternativene inkluderer medikamenter og kirurgi.
I noen tilfeller er symptomene mild, og man kan vente å se om behandling er nødvendig. En sjelden gang kan forandringene gå tilbake av seg selv.
Kortikosteroider (som prednisolon) er førstelinjebehandling for de fleste pasienter. De er effektive i 93% av tilfellene og kan gi en fullstendig tilbakegang av symptomer og funn hos ca. 66%%. Behandlingsregimet er vanligvis prednison 0.6 mg/kg/dag initialt, med gradvis reduksjon med 5 mg hver annen uke (Masaki Y, 2017).
Responsen på behandlingen måles ved kliniske, biokjemiske og radiologiske parametere. Det er viktig å være oppmerksom på risikoen for steroid-diabetes ved bruk av kortikosteroider.
Begynnende tilbakegang av svulster forventes 2-4 uker fra behandlingsstart. Behandlingen bør fortsette i minst 3-6 måneder. I løpet av denne tiden vil svulsten vanligvis ikke lenger komprimere omliggende organer og strukturer.
Det er imidlertid viktig å være klar over at ca. 50% av pasientene får gradvis tilbakefall etter 6 måneder. I praksis kan dosen for eksempel være Prednisolon 30-40mg i inntil 4 uker, deretter nedtrappende doser, med vurdering av seponering etter 2-6 måneder. Residiv-raten er imidlertid 10 til over 50%. Vedvarende behandling reduserer residivene, og målet er å finne lavest mulig effektive dose (for eksempel Prednisolon 2,5-5mg/dag) (Nambiar S, 2021).
Ved store aortaaneurismer, kan kortikosteroider fremprovosere ruptur. Behandlingsnytte må derfor nøye veies opp mot risikoen i hvert enkelt tilfelle (Onkaramurthy NJ, 2023).
Rituksimab er et biologisk legemiddel som hemmer B-celler og har vist effekt i behandlingen av IgG4-relaterte sykdommer. Det er et alternativ (og et mulig supplement) til kortikosteroider, men brukes . som utprøvende behandling /off-label for denne indikasjonen. Doseringen er som ved revmatoid artritt (to doser med 1000mg iv med to ukers mellomrom). Behov for ev. vedlikeholdsbehandling (for eksempel 500 eller 1000mg hver 6 måned eller lengre intervaller) vurderes i hvert enket tilfelle. Behandlingsresponsen forklares, i alle fall delvis, ved at rituksimab interferer med tilbakekomst av kort-levde plasmaceller som produserer IgG4 (Khosroshahi A, 2012).
Dupilumab er et annet biologisk medikament i form av et monoklonalt antistoff. Det virker på interleukin 4 (IL-4)-reseptor alfa, en reseptor som deles av IL-4 og IL-13. Fysiologisk påvirker IL-4 at immunsystemet bytter fra å produsere IgM-antistoffer til IgG4-antistoffer, mens IL-13 er involvert i dannelsen av arrvev (fibrose). Dupilumab som en IgG4-hemmer har derfor potensiale for å være en ny steroidbesparende behandling for IgG4-relatert sykdom (IgG4-RD) (Simpson RS, 2019). Rapporter tyder på en steroidsparende effekt (Kanda M, 2023).
En begrensning ved dupilumab er at det kun hemmer Th2-celler, en type immuncelle.
Andre DMARDs som også brukes er azathioprin (Imurel) (2 mg/kg /dag), mykofenolat mofetil (opp til 2.5 g/dag hvis toleranse), og metotreksat. Dokumentasjon fra randomiserte kontrollerte studier mangler imidlertid for disse legemidlene (Nambiar S, 2021).
Kirurgisk behandling og stenting kan være aktuelt mot obstruksjon forårsaket av pankreas- galle- eller nyre-affeksjon. Karkirurgi kan være nødvendig ved truende aneurismeruptur eller alvorlige stenoser.
Oppfølging
IgG4-RD utvikler seg vanligvis langsomt over måneder eller år. Medikamentell behandling forventes å stanse sykdomsprogresjonen og gradvis redusere størrelsen på de godartede svulstene. Regelmessig klinisk undersøkelse og bildediagnostikk av de affiserte organene er viktig for å overvåke sykdomsaktiviteten. Kontroll av organfunksjonsprøver (f. eks. ALP, bilirubin, kreatinin, urin stiks), og IgG4-nivå i blodprøver er også nødvendig.
Risiko for tilbakefall
Residiv (tilbakefall) av IgG4-RD er relativt vanlig, og derfor anbefales livslang oppfølging. Faktorer som øker risikoen for residiv inkluderer:
- Høye IgG4-nivåer i blodet
- Affeksjon av flere organer
- Atopi (allergisk disposisjon)
- Perifer eosinofili (økt antall eosinofile granulocytter i blodet)
- Høyt IgE-nivå i blodet
Regelmessig oppfølging og monitorering av disse risikofaktorene er viktig for å kunne oppdage og behandle tilbakefall tidlig (Wallace ZS, 2024).
Retningslinjer
Europeiske (UEG og SGF): Löhr J-M, 2020
2019 ACR/EULAR klassifikasjonskriterier for IgG4 relaterte sykdommer (Wallace ZS, 2019)
Internasjonale: Khosroshahi A, 2015
Norsk revmatologisk Forening (prosedyrer)
