BASISKOMPETANSE (REV 001-005)
3 Immunsystemets oppbygging og funksjon, immunologi. Autoimmune sykdommer, autoimmunitet og inflammasjon (REV 003)
Immunsystemet oppbygning
Jan Tore Gran and Øyvind Palm
Læringsmål REV 003. Revmatologen skal ha god kunnskap om immunsystemets oppbygning og funksjon, herunder osteoimmunologi, mekanismer for toleranse og autoimmunitet.
Innledning
Revmatologens viktigste oppgave er å håndtere inflammatoriske og autoimmune sykdommer der kroppens eget immunforsvar (immunsystemet) ved en feil angriper egne vev. Vevene som kan skades er blant annet ledd med synovialmembran og brusk, bindevev, blodkar, hud, nyrer, hjerte og andre organer. Forståelsen av immunsystemets normale funksjon har vist seg å være viktig for å forstå, patogenesen til disse sykdommene, men også prinsippene for moderne behandling (Yatim KM, 2015).
Osteoimmunologi (iht. læringsmålet REV 003) er omtalt her i form av benmargens leukocyttproduksjon med cytokiner etc, mens osteoklast- og osteoblast-funksjonene er beskrevet i kapitlet om osteopoose.
Målet for dette kapitlet er
- Byggestener i immunsystemet. Beskrive komponenter i immunsystemet, slik som celler, molekyler og organer og hvordan de fungerer sammen
- Immunologisk toleranse og autoimmunitet. Forklare hvordan toleranse og autoreaktivitet oppstår og fungerer
- Immunologisk patogenese. Vise hvordan disse mekanismene svikter ved revmatiske sykdommer
- Klinisk betydning. Gi innledende informasjon om klinisk relevans med behandlingsstrategier baserte på immunologi
1. Immunsystemets komponenter og grunnleggende funksjon
Medfødt og adaptivt immunsystem
Oversikt over inndeling i medfødt og adaptiv immunitet (det innate og det adaptive immunsystemet). De enkelte komponenter er nærmere beskrevet nedenfor.
| Egenskap | Medfødt immunitet (innat) | Adaptiv immunitet |
|---|---|---|
| Tidlig respons | Umiddelbar eller innen timer | Tar dager – krever antigenpresentasjon, klonal ekspansjon |
| Spesifisitet | Ikke-spesifikk; mønstergjenkjenning (PAMPs/Pathogen-Associated Molecular Patterns, DAMPs/Damage-Associated Molecular Patterns) | Høy spesifisitet via T- og B-celle reseptorer, antistoffer |
| Hukommelse | Generelt ingen hukommelse (noen moduler‐effekter) | Immunologisk hukommelse; kraftigere respons ved re-eksponering |
| Hovedkomponenter | Barriereforsvar (hud, slimhinner), fagocytter (makrofager, nøytrofile), dendrittiske celler, NK-celler, komplement, inflammasjonsmediatorer | T-lymfocytter (CD4+, CD8+), B-lymfocytter / plasmaceller, antistoffer, hukommelsesceller |
Medfødt immunitet (det innate immunsystemet) (Janeway Jr, TR, 2002)
Dette er den første, raske responsen som ikke er spesifikk for et bestemt agens. Det innate immunsystemet er spredt over hele kroppen. Det består av følgende:
- Beskyttende barrierer: Fysiske barrierer som hud, slimhinner, mucus, ciliebevegelse og kjemiske barrierer som magesyre, enzymer og antimikrobielle peptider.
- Fagocytose: nøytrofile granulocytter, makrofager. Disse cellene gjenkjenner og fjerner patogener ved fagocytose, frigjøring av toksiske substanser og initiering av inflammasjon.
- Myeloide progenitorceller: Disse er kilden til cellene i det innate immunsystemet; nøytrofile-, eosinofile- og basofile granulocytter, mastceller, makrofager, monocytter (som differensierer til makrofager i vev), dendrittiske celler og innate lymfoide celler (inkludert NK-celler).
- Dendrittiske celler: fungere som bro mellom innate og adaptive immunitet ved å oppfange antigen, prosessere og presentere via MHC-molekyler til T-celler.
- NK-celler: dreper virusinfiserte eller stressede celler uten behov for antigenpresentasjon.
- Humorale komponenter: Komplementsystemet, akuttfaseproteiner som C-reaktivt protein (CRP), cytokiner (f. eks. interferon type I (IFN-α/β), interleukin-1 (IL-1) og tumornekrosefaktor alfa (TNF-α). Disse komponentene bidrar til inflammasjon, opsonisering (merking av patogener for fagocytose) og direkte drap av patogener.
- Aktivering via patogenassosierte (fremmede) molekylære mønstre (PAMPs) og faresignaler (DAMPs; kroppsegne molekyler som blir patogene hvis de ved nekrose forlater cellene) via reseptorer som Toll-like reseptorer (TLRs) og NOD-like reseptorer (NLRs).
- Uendret respons: Responsen er relativt uendret ved gjentatt eksponering for samme agens.
Det innate immunsystemet består ikke bare av “kjemiske deler”, men av et samspill mellom cellulære og humorale komponenter. Interferoner er en gruppe cytokiner med antiviral aktivitet, mens IL-1 er et proinflammatorisk cytokin. Sammen med andre cytokiner og komponenter i det innate immunsystemet, kan de utløse kraftige immunreaksjoner og inflammasjon, noe en ser ved autoinflammatoriske (feber-) sykdommer og ved systemisk juvenil idiopatisk artritt (sJIA).
Adaptiv immunitet (det adaptive immunsystemet)
Det adaptive immunsystemet er hovedsakelig lokalisert i de sekundære lymfoide organene (lymfeknuter, milt og Mucosa-Associated Lymphoid Tissue/MALT) hvor immuncellene lærer å gjenkjenne spesifikke patogener, men det utvikles i de primære lymfoide organene (benmarg og tymus).
Funksjonen til det adaptive immunsystemet er å være et spesifikt forsvar ved å gjenkjenne spesifikke antigener (hukommelse). Dette resulterer i en raskere og kraftigere respons ved re-eksponering for samme antigen. Det tar imidlertid noe tid (ofte 7-10 dager) for å utvikle full effekt, men gir til gjengjeld en kraftigere og mer målrettet respons enn det innate immunsystemet.
Lymfocytter: T-lymfocytter (T-celler) og B-lymfocytter (B-celler) er viktige komponenter i det adaptive immunsystemet. Disse cellene utvikles fra hematopoietiske stamceller i benmargen. B-celler modnes i benmargen, mens T-celler modnes i tymus.
-
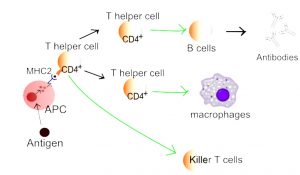
T-celler: Gjenkjenner antigener presentert av antigenpresenterende celler (APC) som dendrittiske celler og makrofager via T-cellereseptoren (TCR) (Sun L, 2023). Det finnes flere undergrupper av T-celler:
- – CD4+ (“hjelper” – Th1, Th2, Th17, T-reg (regulatoriske T-celler) osv.) som koordinerer immunrespons ved å produsere cytokiner og hjelpe B-celler.
- T hjelper 1 (Th1) celler: Produserer hovedsakelig interferon-gamma (IFN-γ), som aktiverer makrofager og er viktig for intracellulær patogenbekjempelse og cellemediert immunitet.
- T hjelper 2 (Th2) celler: Produserer interleukin-4 (IL-4), IL-5 og IL-13, som er viktige for B-celleaktivering, antistoffproduksjon (spesielt IgE) og bekjempelse av ekstracellulære parasitter.
- T hjelper 17 (Th17) celler: Produserer IL-17, som er involvert i inflammasjon og beskyttelse mot ekstracellulære bakterier og sopp, men også spiller en rolle i patogenesen av autoimmune sykdommer.
- Regulatoriske T-celler (Tregs): Produserer immunsuppressive cytokiner som IL-10 og TGF-β, som bidrar til å dempe og kontrollere immunresponsen og opprettholde immuntoleranse (Burzyn D, 2013).
- Follikulære T-hjelpeceller (Tfh): Befinner seg i B-cellefollikler i lymfeknutene og bidrar til B-celleaktivering, klasseskifte av antistoffer og dannelse av langlivede plasmaceller og hukommelses-B-celler.
- – CD8+ (“cytotoksiske”) som dreper celler som presenterer fremmede antigener som virusinfiserte celler, tumorceller og andre skadede celler via MHC I (Major Histocompatibility Complex I) ved å frigjøre cytotoksiske partikler.
- – CD4+ (“hjelper” – Th1, Th2, Th17, T-reg (regulatoriske T-celler) osv.) som koordinerer immunrespons ved å produsere cytokiner og hjelpe B-celler.
-
B-celler og antistoffer
– Differensiering til plasmaceller –> produksjon av antistoffer (immunglobuliner: IgM, IgG, IgA, IgE, IgD)
– Antistoffenes funksjoner: nøytralisering, opsonisering, komplementaktivering, dannelse av immunkomplekser- B-celler gjenkjenner antigener via B-cellereseptoren (BCR) og differensieres til plasmaceller som produserer antistoffer (immunglobuliner). Antistoffer binder seg spesifikt til antigener, nøytraliserer dem og opsoniserer dem for fagocytose eller aktiverer komplementsystemet. B-celler har overflatereseptorer som CD19, CD20, CD22 og BAFF-reseptoren (BAFFR) som er mål for biologisk behandling ved autoimmune sykdommer (Wu F, 2021).
- Antigenpresenterende celler (APC) er dendrittiske celler, makrofager og B-celler. Disse cellene tar opp antigener, prosesserer dem og presenterer dem for T-celler via MHC-molekyler.
Forskjeller mellom det innate og det adaptive immunsystemet
| Egenskap | Innate immunsystem | Adaptive immunsystem |
| Spesifisitet | Begrenset, gjenkjenner PAMPs og DAMPs | Høy, gjenkjenner spesifikke antigener |
| Hukommelse | Ingen | Ja |
| Responstid | Minutter/timer | Dager |
| Hovedkomponenter | Fagocytter (makrofager, nøytrofile), NK-celler, dendrittiske celler, komplementsystemet | Lymfocytter (B- og T-celler), antistoffer |
| Tabell. Man kan dele en del sykdommer inn etter om de hovedsakelig aktiveres av det innate immunsystemet (autoinflammatorisk) eller det adaptive immunsystemet (autoimmune sykdommer) (Shimuzu J, 2023). | |
| Familiær middelhavsfeber (FMF), TRAPS | Autoinflammatoriske. Sjeldne monogene sykdommer/febersyndromer |
| Crohns sykdom (CD), ulcerøs kolitt (UC) | Autoinflammatoriske polygenetiske sykdommer |
| Behcets sykdom, ankyloserende spondylitt/Bekhterevs | Blandet autoinflammatorisk og autoimmunt mønster |
| Revmatoid artritt, systemisk lupus (SLE) | Klassiske autoimmune sykdommer |
| Autoimmunt lymfoproliferativt syndrom (ALPS), immun dysregulert, polyendokrinopati enteropati, X-bundet syndrom (IPEX) | Autoimmune, sjeldne monogene sykdommer |
Lymfoide organer og vev
-
Primære lymfoide organer
– Beinmarg: stamceller, B-celle modning, generering av myeloide og lymfoide celler
– Tymus: T-celle modning, utdanning i toleranse (positiv og negativ seleksjon). -
Sekundære lymfoide organer / perifert vev
– Lymfeknuter: filtrering, møteplass for antigen, APCs, T- og B-celler
– Milt: filtrering av blod, respons mot blodbårne antigener.
– Mukosaassosiert lymfoid vev (MALT): f. eks. i tarm (GALT), luftveier – viktig som “inngangsporter” for patogener.-
I lymfeknuten møter den ferdigutviklede immuncellen (dendrittisk celle) T-cellene. T-celler har T-cellereseptorer (TCRs) som gjenkjenner spesifikke peptider bundet til MHC-molekyler. En naiv CD4+ T-celle aktiveres når dens TCR gjenkjenner et peptid-MHC klasse II-kompleks og mottar kostimulering via interaksjon mellom CD28 på T-cellen og CD80/86 på den dendrittiske cellen. Når T-cellene blir aktivert, skjer det en massiv opprustning i immunforsvaret: De formerer seg raskt og spesialiserer seg til kampklare effektor-T-celler. De lager også hukommelsesceller som sikrer at immunforsvaret husker og raskt kan kjenne igjen inntrengeren ved neste møte. Aktiverte CD4+ T-celler produserer også cytokiner som regulerer immunresponsen ved å aktivere andre immunceller som B-celler, makrofager og endotelceller..
B-celler og antistoffproduksjon: I lymfeknuten kommuniserer B-celler med antigener og Tfh-celler. B-celler har B-cellereseptorer (BCRs) som binder spesifikke antigener. Binding av antigen til BCR og samspill med Tfh-celler (som gir medstimulering via CD40-CD40L interaksjon og cytokiner) aktiverer B-celler. Aktiverte B-celler prolifererer, differensieres til plasmaceller som produserer antistoffer og danner hukommelses-B-celler. Ved den primære immunresponsen produseres hovedsakelig IgM-antistoffer.
Hukommelsesrespons: Hvis en eksponeres igjen for det samme antigenet, aktiveres hukommelses-B-celler raskt. Disse cellene gjennomgår raskere proliferasjon og differensiering til plasmaceller som produserer antistoffer som bedre og sterkere kan binde seg til smittestoffet. I tillegg skjer klasseskifte (isotype switching) av antistoffer der IgM erstattes av IgG, IgA eller IgE, avhengig av cytokinmiljøet og T-cellehjelpen. Hver antistoffisotop har spesifikke funksjoner: IgG kan gjøre patogener lettere gjenkjennelig (opsonisering) og aktivere komplementsystemet. IgA finnes i slimhinner og nøytraliserer patogener der, mens IgE er involvert i type I hypersensitivitetsreaksjoner (allergi) ved å binde til mastceller og frigjøre granula-innhold.
-
Molekylære komponenter og signalstoffer

-Antistoffer (immunglobuliner)
Immunglobuliner (antistoffer) er glykoproteiner produsert av plasmaceller som er differensierte B-lymfocytter. Immunglobuliner er viktige deler av det adaptive immunsystemet og kritiske for humoral immunitet. Immunglobuliner gjenkjenner antigener spesifikt og starter mekanismer som fører til nøytralisering og eliminering av disse. I revmatologi er kunnskap om immunglobuliner viktig for forståelsen av patogenesen ved autoimmune sykdommer og for bruk av immunglobulinbaserte terapier.
Immunglobulinenes funksjon:
- Antigengjenkjenning: Immunglobuliner kan feste seg til spesifikke molekyler på overflaten av antigener via sine variable regioner (Fab-fragmenter). Denne bindingen er grunnlaget for antistoffenes spesifisitet.
- Nøytralisering: Immunglobuliner/antistoffer kan nøytralisere patogener ved å blokkere deres evne til å binde seg til vertsceller eller ved å inaktivere toksiner.
- Opsonisering: Binding av immunglobuliner/antistoffer til patogener øker fagocyttenes evne til å gjenkjenne og fortære dem.
- Komplementaktivering: Visse immunglobulinklasser (IgM og IgG) kan aktivere komplementsystemet med en kaskade som fører til destruksjon av patogener, inflammasjon og opsonisering (binding som tilrettelegger for fagocytose).
- Antistoffavhengig cellulær cytotoksisitet (ADCC): Immunglobuliner/antistoffer bundet til målceller kan rekruttere immunceller som NK-celler og makrofager for å drepe målcellene.
- Immunologisk hukommelse: Etter eksponering for et antigen genereres hukommelses-B-celler som raskt kan differensiere til plasmaceller og produsere store mengder antistoffer ved re-eksponering. Dette er grunnlaget for immunologisk hukommelse og vaksinasjon.
–Immunglobulin-klasser: IgM (første respons), IgG (langvarig), IgA (mucosa), IgE (allergi/parasit), IgD.
- -IgM er den første typen antistoff under en primær immunrespons, f. eks. ved en infeksjon. De er effektive ved aktivering av komplementsystemet og dannes før IgG. Fertile kvinner har generelt noe høyere IgM-nivåer. Ved høy IgM skilles mellom en polyklonal, oligoklonal og monoklonal økning. Høye nivåer av IgM kan indikere en nylig infeksjon. Poly- eller oligoklonal høy IgM kan ses også ved leversykdommer og autoimmune sykdommer. Monoklonal økning kan gi mistanke om makroglobulinemi (Waldenstöms), AA amyloidose og MUGS (monoklonal udefinert gammopati av usikker signifikans). Lav IgM ses ved primær og sekundær immunsvikt.
- -IgG er det viktigste og vanligste immunglobulinet i beskyttelse mot infeksjoner. Det utgjør 75-80% av de sirkulerende immunglobulinene og har en halveringstid på 23 døgn. IgG dannes senere i sykdomsforløpet enn IgM ved infeksjoner. Det er viktig i sekundær immunrespons og gir langvarig beskyttelse. IgG kan i svangerskap passere placenta og beskytte fosteret. Mange TNF-hemmere (biologiske legemidler) som brukes mot revmatoid artritt og andre revmatiske sykdommer består av IgG. Polyklonalt forhøyede verdier ses ved Sjøgrens sykdom. IgG inndeles i subklasser IgG1, IgG2, IgG3, IgG4 med flere som har ulike funksjoner (Salmon JE, 2001). Ved IgG4 relaterte sykdommer er IgG4 ofte forhøyet i serum eller i vevet. Mange terapeutiske antistoffer (f. eks. TNF-hemmere) er IgG-baserte. Behandling med det biologiske legemiddelet rituksimab (mot revmatoid artritt, ANCA-vaskulitt som GPA, MPA, EGPA og andre) kan redusere nivåetg av IgG og øke infeksjonsrisikoen. Monoklonal IgG ses i serum-elektroforese ved MUGS (se nedenfor) og myelomatose.
- -IgA finnes hovedsakelig i slimhinner (mucosa) og kroppsvæsker som spytt, tårer, melk og sekreter fra mage-tarmkanalen og luftveiene. IgA beskytter mot patogener på slimhinneoverflater. IgA kan avleires i nyrene ved IgA-nefropati. IgA-vaskulitt (Henoch-Schönleins purpura) involverer IgA-avleiringer i blodårer, hud, ledd og nyrer. IgA mangel ses hos 0,3% av befolkningen og er den vanligste primære immunsvikten. Den disponerer blant annet for alvorlig forløp av systemisk lupus (SLE), men de fleste med IgA-mangel er uten symptomer (Killeen RB, 2023 sp).
- -IgE er involvert i type I hypersensitivitetsreaksjoner. Det binder seg til mastceller og basofile granulocytter. Ved binding av antigen frigjøres mediatorer som histamin som forårsaker allergiske symptomer. Økte IgE-nivåer sees ved allergiske reaksjoner, parasitt-sykdommer og enkelte revmatiske sykdommer slik som EGPA (eosinofil granulomatose med polyangiitt/Churg-Strauss vaskulitt og eosinofil fasciitt).
- -IgD: Funksjonen er ikke fullstendig klarlagt. Det finnes hovedsakelig på overflaten av naive B-celler og antas å spille en rolle i B-celleaktivering.
Paraproteiner, monoklonale komponenter og MUGS.
Monoklonale gammopatier karakteriseres av produksjon av et monoklonalt immunglobulin (paraprotein) av en enkelt plasmacelleklon.
Hos ca. 2% av alle personer over 50 års alder påvises monoklonal gammopati. De fleste tilfellene er ikke assosiert med sykdomstegn og er av usikker betydning (Monoclonal gammopathy of undetermined significance = MUGS) (Ørstavik R, 2002). Ved MUGS er mengden paraprotein mindre enn 30 g/L i serum, og plasmacellene utgjør mindre enn 10% i benmargen. Man påviser ikke tegn til organskader (CRAB-kriterier: hyperkalsemi, nyresvikt, anemi, benlesjoner), og det er ikke tegn til annen B-celle proliferativ sykdom/lymfom. MGUS har lav risiko for progresjon til myelomatose eller andre maligne plasmacellesykdommer, men årlig kontroll med serumproteinelektroforese anbefales. Dersom mengden monoklonalt protein øker, anbefales henvisning til spesialist i blodsykdommer (hematolog).
")
-Cytokiner, kemokiner, vekstfaktorer
Cytokiner er en gruppe signalproteiner som spiller en avgjørende rolle i intercellulær kommunikasjon og regulering av immunresponser. De kan ha pro-inflammatoriske eller anti-inflammatoriske effekter. Hemming av pro-inflammatoriske cytokiner er en sentral strategi i behandlingen av inflammatoriske revmatiske sykdommer, f. eks. ved bruk av biologiske legemidler som TNF-hemmere og IL-6-reseptorantagonister (Liu S, 2022).
–Proinflammatoriske cytokiner:
Proinflammatoriske cytokiner produseres av immunceller, inkludert makrofager, dendrittiske celler, T-hjelpeceller (spesielt Th1 og Th17) og andre som fibroblaster og endotelceller. Når det innate immunsystemet ikke klarer å eliminere den utløsende årsaken, forsterkes inflammasjonen gjennom økt produksjon av pro-inflammatoriske cytokiner, noe som aktiverer det adaptive immunsystemet. Dendrittiske celler som brobyggere mellom det innate og adaptive immunsystemet presenterer antigener for naive T-celler og gir kostimulerende signaler for å initiere en adaptiv immunrespons. En hensiktsmessig inflammasjon er en viktig del av kroppens forsvarsmekanisme, men vedvarende eller dysregulert inflammasjon kan føre til vevsskade og kroniske sykdommer.
- TNF-α (Et kraftig pro-inflammatorisk cytokin som induserer inflammasjon, apoptose og systemiske effekter), IL-1 (Induserer inflammasjon, feber og akuttfaserespons),
- IL-6 (Stimulerer produksjon av akuttfaseproteiner i leveren og bidrar til B-celle differensiering.),
- IL-17 (Involvert i rekruttering av nøytrofile granulocytter og patogenesen av flere autoimmune sykdommer.), interferon-γ (Aktiverer makrofager og fremmer Th1-responser.).
- IL-12 (Øker differensiering av naive T-celler til Th1-celler og produksjon av IFN-γ). Granulocytt-makrofag koloni-stimulerende faktor (GM-CSF) stimulerer produksjon og differensiering av granulocytter og makrofager.
- IL-15, IL-18 og IL-23 viktige i inflammasjonsprosesser. IL-23 spiller en sentral rolle i Th17-responsen (Liu S, 2022).
– Anti-inflammatoriske/regulatoriske cytokiner: Disse cytokinene demper immunresponsen og bidrar til å opprettholde immunologisk homeostase. Viktige anti-inflammatoriske cytokiner inkluderer:
- Interleukin-10 (IL-10): Hemmer produksjonen av pro-inflammatoriske cytokiner og demper T-celleaktivering.
- Transforming growth factor beta (TGF-β): Har immunsuppressive effekter og bidrar til vevsreparasjon og fibrose.
- Interleukin-4 (IL-4): Fremmer Th2-responser og B-celle differensiering til plasmaceller.
- IL-35: Utskilles av celler og hemmer og immunresponser for å dempe inflammasjon og øke immunologisk toleranse.
– Kemokiner bidrar til å trekke immunceller til betennelsessteder.
| Egenskap | Kemokiner | Cytokiner |
| Klassifisering | Spesialisert undergruppe | Paraplybegrep (bred kategori) |
| Hovedfunksjon | Tiltrekke immunceller (kemotaksi). | Regulere immuncellenes funksjon (vekst, aktivering) |
| Primær effekt | Bevegelse av celler. | Signalering mellom celler (stimulering/hemming). |
| Eksempler | CCL2, CXCL8 | (og kemokiner er inkludert). |
-Komplement
– Kaskadesystem som er viktig for opsonisering av patogener, lyse av celler og modulering av inflammasjon.
Komplementsystemet er spesielt viktig for det innate immunsystemet, men spiller også en viktig rolle i det adaptive immunsystemet. Det består av en kaskade av plasmaproteiner som aktiveres i en sekvensiell reaksjon (kaskadeaktivering). Komplementsystemet er avgjørende for å fjerne patogener, men ukontrollert eller uhensiktsmessig aktivering kan bidra til vevsskade ved autoimmune inflammatoriske sykdommer.
Komplementsystemets funksjon: Komplementsystemet har flere viktige funksjoner i immunforsvaret:
- Opsonisering: C3b binder seg til overflaten av patogener og “merker” dem for fagocytose, noe som øker effektiviteten av fagocyttenes evne til å gjenkjenne og fortære patogenener.
- Inflammasjon: Aktiveringsprodukter som C3a og C5a (anafylatoksiner) induserer inflammasjon ved å rekruttere immunceller (f. eks. nøytrofile granulocytter) til infeksjonsstedet, øke vaskulær permeabilitet og aktivere mastceller.
- Lyse av patogener: Dannelsen av det terminale komplementkomplekset (TCC)/Membrane Attack Complex (MAC) fører til porer i cellemembranen til patogener og til celledød (lyse).
- Nøytralisering av virus: Noen komplementkomponenter (f. eks. C4b) kan direkte nøytralisere virus ved å forhindre deres evne til å infisere celler.
- Fjerning av immunkomplekser: Komplementsystemet bidrar til å fjerne immunkomplekser (antigen-antistoff-komplekser) fra sirkulasjonen, noe som forhindrer avleiring og vevsskade.
Komplementsystemets aktiveringsveier: Komplementsystemet kan aktiveres via tre hovedveier:
- Den klassiske aktiveringsveien: Utløses av binding av C1q til immunkomplekser (antigen-antistoff-komplekser). C1q binder seg til Fc-delen av IgG eller IgM antistoffer som er bundet til antigen. Dette starter en kaskade av proteolytiske aktiveringer av komplementkomponentene C1r, C1s, C4 og C2 som til slutt fører til dannelse av den klassiske C3-konvertasen (C4b2a).
- Lektinveien: Aktiveres ved binding av mannose-bindende lektin (MBL) eller ficoliner til karbohydrater på overflaten av mikroorganismer. MBL og ficoliner er proteiner som ligner C1q og aktiverer proteaser kalt MASP-1 og MASP-2. Disse proteasene spalter C4 og C2 og danner lektinveiens C3-konvertase (C4b2a) som er identisk med den klassiske C3-konvertasen.
- Den alternative aktiveringsveien: Aktiveres direkte av overflater på mikroorganismer, celleoverflater eller andre substanser uten behov for antistoffer. Denne veien involverer spontan hydrolyse av C3 til C3(H2O) som binder faktor B. Faktor B spaltes deretter av faktor D og danner den alternative C3-konvertasen (C3bBb). Denne konvertasen kan deretter forsterke aktiveringen av veien ved å spalte mer C3.
Alle tre aktiveringsveiene møtes ved dannelsen av C3-konvertase som spalter C3 til C3a og C3b. C3b er sentral i merking/opsonisering og dannelsen av C5-konvertase. C5-konvertasen spalter C5 til C5a og C5b. C5b initierer dannelsen av TCC (MAC).
Komplementsystemet ved autoimmune sykdommer: Ukontrollert eller uhensiktsmessig aktivering av komplementsystemet skjer i patogenesen av flere autoimmune sykdommer:
- Systemisk lupus erythematosus (SLE): Avleiring av immunkomplekser aktiverer den klassiske aktiveringsveien, noe som fører til inflammasjon og vevsskade i ulike organer som nyrer (lupus nefritt), hud, ledd og serøse hinner. Genetiske defekter i komplementkomponenter (spesielt C1q-mangel) er sterkt assosiert med utvikling av SLE. Måling av C3 og C4 kan være nyttig for å vurdere sykdomsaktivitet, da nivåene ofte er lave ved aktiv sykdom på grunn av forbruk.
- Katastrofalt antifosfolipidsyndrom (CAPS): Kjennetegnes av raskt utviklende multiple tromboser i små og store blodårer, ofte assosiert med organsvikt. Aktivering av komplementsystemet bidrar til denne trombosetendensen og organskaden.
- Revmatoid artritt (RA): Komplementaktivering i synovialvæsken i leddene bidrar til inflammasjon og ledd-destruksjon.
- Vaskulitt: Komplementaktivering kan bidra til inflammasjon og skade på blodåreveggen ved ulike former for vaskulitt, inkludert ANCA-vaskulitt.
Måling av komplementaktivering:
- Måling av komplementfaktorer (C3, C4): Reduserte nivåer av C3 og C4 kan indikere forbruk på grunn av aktivering av komplementsystemet, noe som ofte sees ved aktive autoimmune sykdommer.
- Måling av komplementaktiveringsprodukter (f. eks. C3a, C5a, SC5b-9 (TCC)): Direkte måling av aktiveringsprodukter gir mer spesifikk informasjon om komplementaktivering.
Behandling rettet mot komplementsystemet:
- Eculizumab: Et monoklonalt antistoff som hemmer C5 og dermed blokkerer dannelsen av C5a og MAC. Brukes i behandling av tilstander som paroksysmal nattlig hemoglobinuri (PNH), atypisk hemolytisk uremisk syndrom (aHUS) og visse former for myasthenia gravis. Det er også vist effekt ved alvorlig SLE med C1q mangel (som nevnt) (Pickering MC, 2015; Coppo R, 2015).
- Avacopan: En selektiv C5a-reseptorantagonist som blokkerer effekten av C5a. Har vist lovende resultater i behandling av ANCA-vaskulitt i kombinasjon med standardbehandling (Jayne D, 2019).
-Andre effektormekanismer
– Frie radikaler, oksidativt stress, matriksmetalloproteinaser (MMPs) som ødelegger vev.

2. Toleranse og forebygging av autoimmunitet
Immunsystemet har en viktig funksjon i å beskytte kroppen mot fremmede inntrengere. Samtidig må det tolerere kroppens egne antigener (selv-antigener) for å unngå autoimmun reaksjon og angrep på friskt vev. Denne toleransen kalles immuntoleranse eller selvtoleranse (snl.no). Når immuntoleransen svikter, f. eks. ved at immunsystemet reagerer på proteiner som ligner på kroppens egne (kryssreaktive autoantigener, molecular mimicry), forstyrres likevekten mellom selvtoleranse og autoaggresjon. Dette kan føre til at immunsystemet aktiveres og ved en feil angriper friskt vev. Det kan forklare hvorfor noen autoimmune sykdommer affiserer flere organsystemer (Yatim KM, 2015).
Immunsystemet har kompliserte kontrollmekanismer som gjør at celler som reagerer mot kroppseget (“selvantigener”) enten fjernes, undertrykkes eller inaktiveres. Disse mekanismene kan deles inn i sentral og perifer toleranse (Long A, 2023).
Betydningen av oppdagelsen av immuntoleranse ble illustrert ved tildelingen av Nobelprisen i fysiologi eller medisin for 2025 (Mary E. Brunkow, Fred Ramsdell og Shimon Sakaguchi).
Sentral toleranse
-
T-celler i thymus (T for thymus) gjennomgår “positiv seleksjon” (de som kan binde MHC) og “negativ seleksjon” (de som binder selvantigener med høy affinitet → apoptose).
-
B-celler i benmarg (B for benmarg): kan gjennomgå klonal sletting, receptor editing (endre antigenreseptor hvis autoreaktivitet oppdages) eller apoptose.
Genetiske mekanismer viktige her, f.eks. AIRE (Autoimmune Regulator)-transkripsjonsfaktor som gjør at thymus uttrykker perifer vevsantigener og kan “trene” T-celler til ikke å reagere mot disse. Forstyrrelser i AIRE fører til autoimmun syndrom.
Den viktigste mekanismen for sentral T-celle toleranse er negativ seleksjon i thymus. Under T-cellemodningen i thymus presenteres selv-peptider bundet til MHC-molekyler for thymocytter (umodne T-celler). Thymocytter som binder seg for sterkt til selv-peptider gjennomgår apoptose (programmert celledød) og elimineres. Denne prosessen fjerner de fleste autoreaktive T-celler før de forlater thymus. I tillegg kan noen thymocytter som binder selv-antigener med moderat affinitet, differensiere til regulatoriske T-celler (Tregs) i thymus (thymisk Tregs eller tTregs) (Sprent J, 2005).
Perifer toleranse
Når noen autoreaktive lymfocytter unnslipper sentral toleranse, finnes det flere mekanismer i periferien:
-
Regulatoriske T-celler (Tregs): Undertrykker aktivering av effektor T‐celler, produserer antiinflammatoriske cytokiner (IL-10, TGF-β). Tregs er avgjørende for å opprettholde perifer toleranse og forhindre autoimmunitet.
-
Anergi: Hvis en T-celle møtes med antigen uten co-stimulering (f.eks. via CD28-CD80/86 interaksjon), blir den “gjemt” eller ikke-reaktiv.
-
Apoptose / aktiveringsindusert celle død: spesielt ved overaktivering. Aktiverte T-celler cellene blir fjernet etter at de har utført jobben sin (aktiveringsindusert celledød/AICD), noe som bidrar til å begrense immunresponsen.
- Definisjon. Ordet apoptose kommer fra gresk og karakteriserer kronbladenes fall fra blomsten. Apoptose fører til organisert ødeleggelse av celler (programmert celledød). Prosessen er i utgangspunktet fysiologisk og et viktig ledd i balansen mellom celleproliferasjon og celledød. En antar at 90 % av cellene dør i apoptose, mens resten dør av nekrose (celleskade). Til forskjell fra nekrose gir apoptose lite utslipp av intracellulært materiale, og prosessen ledsages ikke av inflammasjon. Tvert imot, apoptoptiske celler kan frigjøre antiinflammatoriske metabolitter (via panneksin 1-kanaler) (Medina CB, 2020).Patogenese. Apoptose i en celle starter ofte ved at Fas (“døds-reseptor”) på overflaten av cellen bindes til sin ligand Fas (FasL). Bindingen mellom Fas og FasL fører til aktivering av flere intracellulære signalveier, bl.a. en som involverer aktivering av ulike kaspaser. Kaspasene aktiverer endonukleaser (DNAser) og stimulerer til endringer av celleoverflaten. Kaspasene påvirkes av celledød-faktorer, fravær av overlevelses-faktorer, celleskade og skade på DNA. Ved ikke-reparativ skade av DNA vil p53 (genomets vokter) indusere dannelse av heterodimeren bcl-bax med apoptose av cellen til følge. BcI2 som er lokalisert til mitokondriemembranen hemmer kaspasene og beskytter dermed mot apoptose. IL-1, TGF-beta og alfa- hemmer også apoptose. Fas-antigenet er et overflateprotein som tilhører TNF familien og er viktig for destruksjon av aktiverte T-Iymfocytter.Ved SLE er det påvist økte mengder celler i apoptose i perifert blod. Dette er sannsynligvis et tegn på nedsatt evne til å fjerne slike celler. Hvis celler isteden destrueres ukontrollert, kan intracellulære antigener frigis. Mot slike antigener er det ikke utviklet immunologisk toleranse. På denne måten kan autoimmunitet utløses.
-
Antigenignoranse/ignorering/immunologisk ignorans: Visse antigener er “skjult” eller ikke tilgjengelig for immunovervåkning under normale forhold. Autoreaktive lymfocytter kan ignorere selv-antigener hvis disse er utilgjengelige eller presenteres i lave konsentrasjon.
Faktorer som kan bryte toleranse
Etiologien til autoimmune sykdommer er multifaktoriell og involverer et komplekst samspill mellom genetisk predisposisjon og miljøfaktorer. Konkordansrater blant monozygote (eneggede) tvillinger varierer mellom 12 og 67 %, noe som understreker viktigheten av både genetiske og miljømessige bidrag (Wang L, 2015).
-
Genetisk predisposisjon: varianter i HLA-klasse II (og I), genmutasjoner i gener som regulerer cytokiner, signalveier, regulatorceller.
- Polyautoimmunitet defineres som samtidig forekomst av to eller flere autoimmune sykdommer hos samme individ. Autoimmun tyreoiditt er ofte en del av dette bildet (Bliddal S, 2017).
- Multiple autoimmune syndromer (MAS): Defineres ved koeksistens av tre eller flere autoimmune sykdommer hos samme individ. Genetisk predisposisjon, spesielt knyttet til HLA klasse I og II, og kjønn (kvinner er mer utsatt), samt miljøfaktorer som røyking og sammensetningen av tarmens mikrobiota spiller en rolle i utviklingen av MAS. De vanligste kombinasjonene inkluderer autoimmun thyreoiditt og Sjøgrens syndrom (Roja-Villarraga A, 2012).
- Autoimmune sykdommer rammer flest kvinner (omtrent 80 %). Denne kvinne-dominansen kan skyldes hormoner (østrogen og progesteron kan modulere immunresponsen), forskjeller i immuncellenes funksjon mellom kjønnene, men også genetiske faktorer knyttet til X-kromosomet (X-inaktivering og gener som koder for immunrelaterte proteiner) og (Dou DR, 2024).
-
Miljøfaktorer: infeksjoner (molekylær mimicry), vevsskade, eksponering for skjulte antigener, mikrobiom-ubalanse.
-
Dysfunksjon i regulatoriske mekanismer: Treg-svikt, manglende suppresjon, ubalanse mellom effektor og regulatoriske celler.
-
Unødig aktivering av medfødte immunsensorer (PRRs, TLRs), produksjon av cytokiner og kostimulatoriske signaler.
De fleste adaptive immunreaksjoner initieres av at et antigen (fremmed eller selv-antigen) bearbeides av antigenpresenterende celler (APC). APC-er presenterer deretter antigenfragmenter bundet til MHC-molekyler (Major Histocompability Complex) for T-celler. Denne presentasjonen er nødvendig for aktivering av T-celler. De viktigste APC-ene er dendrittiske celler, makrofager og B-celler (snl.no). Dendrittiske celler er de mest potente i å starte primære T-celleresponser.
3. Patogenese ved revmatiske autoimmune sykdommer
Her beskrives hvordan immunologiske feil fører til de typiske trekk i revmatisk autoimmun sykdom.
Initial aktivering og triggerfaser
-
Trigger: Kan være infeksjon, vevsskade, eksponering for skjulte antigener, miljøfaktorer som røyking, hormonell påvirkning.
- Antigen. Et antigen er et stoff som kan bli gjenkjent av immunsystemet og utløser en immunrespons, ofte med produksjon av antistoffer. Antigener kan være proteiner, polysakkarider, lipider eller nukleinsyrer. I forbindelse med autoimmunitet er det kroppens egne molekyler (selv-antigener) som blir målet for immunresponsen.
- For at immunforsvaret skal reagere på et antigen, må antigenet først passere kroppens anatomiske og fysiske barrierer. For eksempel må en tarmpatogen bakterie passere magesyre, mucus (slim) og tarmens epitel før den møter immunsystemets celler.
- Det er få revmatiske sykdommer med kjent antigen: urinsyregikt, kolesterol-embolier, kreft-utløst dermatomyositt, avstøtningsreaksjon (GVHD), infeksjoner som septisk artritt, borreliose og Whipples sykdom.
-
Medfødte celler som “første responder”: dendrittiske celler / makrofager aktiveres, tar opp antigen / DAMPs / PAMPs, modnes og uttrykker kostimulatoriske molekyler.
-
De første cellene som møter antigenet er gjerne celler i det innate immunsystemet med makrofager og dendrittiske celler. Disse cellene har mønstergjenkjenningsreseptorer (PRRs) som Toll-like reseptorer (TLRs), NOD-like reseptorer (NLRs) og RIG-I-like reseptorer (RLRs). Disse reseptorene fungerer som “vakter” i kroppen som gjenkjenner spesifikke, faste kjennetegn på inntrengere. Disse kjennetegnene kalles PAMPs (patogenassosierte molekylære mønstre). PAMPs er som en strekkode eller et passord som finnes på mange smittestoffer, men ikke på kroppens egne celler. Eksempler er lipopolysakkarid (LPS) på utsiden av gramnegative bakterier og peptidoglykan i celleveggen til grampositive bakterier. Ved å gjenkjenne disse faste strukturene vet immunforsvaret umiddelbart at det er en inntrenger de må angripe.
Binding av PAMPs til TLRs aktiverer cellen. Den aktiverte cellen fagocyterer eller endocyterer og bryter ned patogenet, samt produserer cytokiner og kjemokiner. Cytokinene starter inflammasjon, rekrutterer andre leukocytter til infeksjonsstedet og bidrar til aktivering av det adaptive immunsystemet.
-
-
Antigenpresentasjon til naive T-celler → aktivering av effektor T-celler (Th1, Th17) → produksjon av cytokiner som IL-17, IFN-γ, TNF-α.
-
Etter aktivering av et patogen gjennomgår dendrittiske celler en modningsprosess. Når cellene utvikler seg og modnes, begynner de å produsere flere MHC-molekyler (Major Histocompatibility Complex), spesielt MHC klasse II, og kostimulerende molekyler (f.eks. CD80/86). Disse MHC-molekylene er som flagg eller utstillingsmontere på overflaten av cellen, og deres oppgave er å vise fram deler av en inntrenger (antigener) til immunforsvaret. Ved å øke antallet MHC-molekyler blir cellene mye bedre til å varsle T-cellene om fare.
Den dendrittiske cellen bryter ned antigener fra patogenet til peptider og presenterer disse peptidene bundet til MHC klasse II-molekyler på celleoverflaten. Samtidig migrerer den dendrittiske cellen via lymfatiske kar til nærmeste lymfeknute.
-
Spredning og vedlikehold av immunrespons
-
Epitope spredning: autoimmun respons som opprinnelig er rettet mot et antigen, utvides til å inkludere flere antigener/epitoper.
-
Autoantistoffer: dannes av B-celler/plasmaceller, kan dannes mot modifiserte selvantigener (f.eks. citrullinerte proteiner i RA). Immunkomplekser kan avsettes, aktivere komplement og igangsette inflammasjon.
- Antistoff (immunglobuliner) kan være veldig spesifikke og naturlig utviklede immunglobuliner som binder seg til antigen eller patologiske celler for å fjerne sykdom (se Immunglobuliner, beskrevet ovenfor). Tilstedeværelsen av autoantistoffer betyr ikke nødvendigvis at en person har en autoimmun sykdom. Lave nivå av autoantistoffer kan påvises hos friske individer, spesielt med økende alder og kan forekomme transitorisk ved infeksjoner, vevsskader eller iskemi. Imidlertid er høye titere av spesifikke autoantistoffer ofte et viktig kjennetegn for autoimmune sykdommer (Long A, 2023; Kono H, 2008).
-
Eksempler på autoantistoffer som er relevante i revmatologi inkluderer:
- Anti-dsDNA: Assosiert med SLE.
- Anti-CCP (syklisk citrullinpeptid): Høy spesifisitet for RA.
- Anti-gliadin/anti-transglutaminase: Assosiert med cøliaki.
- Anti-TPO (tyreoideaperoksidase) og TRAb (TSH-reseptor antistoff): Assosiert med autoimmun thyreoiditt.
Noen autoimmune sykdommer ikke har kjente eller lett målbare autoantistoffer (seronegative sykdommer) som nevnt ovenfor. Biologiske legemidler (monoklonale antistoffer) er terapeutisk fremstilte antistoffer som er spesifikke for et enkelt antigen-epitop (en bestemt del av et antigen). De brukes i behandling av en rekke autoimmune sykdommer ved å blokkere eller hemme spesifikke molekyler involvert i immunresponsen.
-
Inflammasjon og vevsskade: Cytokiner aktiverer immunceller, stimulerer produksjon av proteolytiske enzymer (MMPs), oksidativt stress, osteoklasters aktivitet (beinresorpsjon), påvirkning av brusk.
- Definisjon. Inflammasjon er en reaksjon fra organismens side overfor enhver vevsskade. Den har som hovedoppgave å begrense skaden og å sørge for reparasjon. Komplementsystemet og prostaglandiner er viktige via signalstoffer i inflammasjonsprosessen. Ved autoimmune revmatiske sykdommer aktiveres inflammasjonsprosessen ved en feil og vev- og organskader kan oppstå (Long A, 2023). Hensikten med immunsuppressive medikamenter som kortikosteroider, DMARDs, JAK-hemmere, biologiske legemidler er da å dempe eller hemme inflammasjonsprosessen.Akutt inflammasjon. Skade eller sykdom kan nesten umiddelbart utløse en akutt, kortvarig inflammasjon. Smerte, rødhet, redusert funksjon, hevelse og økt varme er klassiske symptomer, men kan være mer eller mindre fraværende (“silent inflammation”). Utmattelse, feber og kvalme kan også foreligge. Akutt inflammasjon varer få dager, subakutt inflammasjon 2-6 uker.Akutt fase reaksjon. Inflammasjonen forårsaker endringer i fordelingen av plasmaproteinene (akutt fase reaksjon), slik en kan se ved serum-elektroforese. De fleste akutt fase proteiner produseres av hepatocytter som stimuleres hovedsakelig av cytokinene interleukin-1 (IL-1) og IL-6 og har en rekke biologiske funksjoner, slik som koagulasjon, fibrinolyse og fagocytose. Følgende substanser øker ved en akutt fase reaksjon: CRP, serum amyloid protein A, fibrinogen, haptoglobin, C3 og gammaglobulin. Albumin og transferrin synker.Inflammatorisk reaksjon kan deles inn i tre faser
- Vaskulær reaksjon
- Eksudativ eller cellulær fase
- Proliferativ (reparasjon) fase
Vaskulær reaksjon. Straks etter skaden eller tilsynekomst av et fremmed agens frigir mastceller og makrofager (det inate immunsystemet) histamin, TNF og IL-1, IL-6. Disse stimulerer utvidelse venuler, deretter arterioler. Samtidig nedsettes gjennomblødningen i skadestedet med følge at blodvæske (plasma) siver ut i det skadede stedet.
Eksudativ eller cellulær fase. Neste skritt er en opphopning av leukocytter på skadestedet. Adhesjonsmolekyler på endotel i blodkar stimuleres slik at neutrofile granulocytter og monocytter fra blodet lettere kan passere gjennom blodåreveggen og infiltrere vevet. Monocyttene i vevet modner til makrofager som sammen med monocyttene fagocyterer og eliminere agens. Også plasmaproteiner som komplement og fibrinogen infiltrerer vevet og angriper patogene agens.
Proliferativ eller reparativ reaksjon. Reparasjonsprosessen skjer ved hjelp av celler som kan nydanne visse strukturer. Angioblaster vil for eksempel kunne danne nye blodkar, mens fibroblaster danner selve grunnsubstansen i arret.
Vaskulogenese er dannelse av kar fra progenitor-celler, angiogenese er dannelse av kar fra preeksisterende kar (“knoppskyting”).
Kronisk inflammasjon skiller seg fra akutt inflammasjon ved sin langvarige natur og karakteriseres av et samspill mellom immunceller, inflammatoriske mediatorer og vevsceller. I motsetning til akutt inflammasjon, som ofte involverer betydelig væskeansamling (ødem), kjennetegnes kronisk inflammasjon av infiltrasjon av mononukleære celler som lymfocytter og makrofager, vevsdestruksjon og samtidig forsøk på reparasjon med fibrose-utvikling (arrdannelse).
Kronisk inflammasjon sees ved autoimmune revmatiske sykdommer som artritt-sykdommer, bindevevssykdommer, vaskulitt, autoinflammatoriske sykdommer. Den spiller en viktig rolle også i utviklingen av aterosklerose som underliggende årsak til hjerte- og karsykdommer, metabolske sykdommer (diabetes og metabolsk syndrom) og nevrodegenerative sykdommer (Alzheimers- og Parkinsons sykdom).
- Definisjon. Inflammasjon er en reaksjon fra organismens side overfor enhver vevsskade. Den har som hovedoppgave å begrense skaden og å sørge for reparasjon. Komplementsystemet og prostaglandiner er viktige via signalstoffer i inflammasjonsprosessen. Ved autoimmune revmatiske sykdommer aktiveres inflammasjonsprosessen ved en feil og vev- og organskader kan oppstå (Long A, 2023). Hensikten med immunsuppressive medikamenter som kortikosteroider, DMARDs, JAK-hemmere, biologiske legemidler er da å dempe eller hemme inflammasjonsprosessen.Akutt inflammasjon. Skade eller sykdom kan nesten umiddelbart utløse en akutt, kortvarig inflammasjon. Smerte, rødhet, redusert funksjon, hevelse og økt varme er klassiske symptomer, men kan være mer eller mindre fraværende (“silent inflammation”). Utmattelse, feber og kvalme kan også foreligge. Akutt inflammasjon varer få dager, subakutt inflammasjon 2-6 uker.Akutt fase reaksjon. Inflammasjonen forårsaker endringer i fordelingen av plasmaproteinene (akutt fase reaksjon), slik en kan se ved serum-elektroforese. De fleste akutt fase proteiner produseres av hepatocytter som stimuleres hovedsakelig av cytokinene interleukin-1 (IL-1) og IL-6 og har en rekke biologiske funksjoner, slik som koagulasjon, fibrinolyse og fagocytose. Følgende substanser øker ved en akutt fase reaksjon: CRP, serum amyloid protein A, fibrinogen, haptoglobin, C3 og gammaglobulin. Albumin og transferrin synker.Inflammatorisk reaksjon kan deles inn i tre faser
Eksempler på sykdomsspesifikke immunmekanismer
| Sykdom | Viktige immunologiske kjennetegn |
|---|---|
| Revmatoid artritt (RA) | Anti-CCP/anti-citrullinerte antistoffer, RF; synovialhyperplasi; invasiv pannus; osteoklastaktivering; rikelig produksjon av TNF, IL-6, Th17-celler |
| Systemisk lupus erythematosus (SLE) | ANA, anti-DNA antistoffer, immunkomplekser, komplementforbruk, multiorgansykdom; type I interferon platform; tap av toleranse i B-celle linjen |
| Spondyloartritter | Entesitt, aksial inflammasjon; IL-17/IL-23 akser; binding til HLA-B27; inflammasjon av sene‐ og ligamentfester; delvis mekanismer med medfødt immunitet og inflammasjon. |
| Bindevevssykdommer generelt | Autoantistoffer (anti-Ro/La, anti-dsDNA, anti-Sm, anti-Scl-70 etc.); vaskulitt; fibrose; påvirkning av regulerende og effektor-celler; cytokinstormer i enkelte sammenhenger |
. CC BY 2,5")
– Neutrophil Extracellular Traps (NETs):
Overdreven eller ineffektiv fjerning av apoptotiske celler/NETs kan eksponere selvantigener og drive inflammasjon. NETs er nettverkslignende strukturer som frigjøres av nøytrofile granulocytter og er en del av immunresponsen. NETs består av et nettverk av løsnet/dekondensert kromatin (DNA og histoner) og granula, proteiner (myeloperoksidase (MPO), proteinase 3 (PR3), elastase og katekpsin G). Disse strukturene fanger og dreper ekstracellulære patogener som bakterier og sopp.
Prosessen der nøytrofile frigjør NETs kalles NETose. Det finnes flere mekanismer for NETose:
- Suicidal NETose involverer aktivering av nøytrofile, etterfulgt av produksjon av reaktive oksygenforbindelser (ROS), aktivering av peptidylarginin deiminase 4 (PAD4) som fører til histon citrullinering og dekondensering av kromatin. Til slutt rupturerer cellemembranen, og NETs frigjøres.
- Vital NETose: Nøytrofile kan frigjøre NETs uten å dø. Dette involverer vesikulær frigjøring av kromatin og granula proteiner.
NETs spiller en viktig rolle i bekjempelsen av infeksjoner, men de er også involvert i patogenesen av flere inflammatoriske og autoimmune sykdommer (He Y, 2018). Mekanismene inkluderer:
- Levering av autoantigener: NETs inneholder en autoantigener som MPO og PR3. Disse kan utløse autoantistoffproduksjon og bidra til utvikling av autoimmune sykdommer som ANCA-vaskulitt. Ved ANCA-vaskulitt er antistoffer mot MPO og PR3 sentrale i patogenesen.
- Forsterkning av inflammasjon: NETs kan aktivere komplementsystemet, indusere produksjon av cytokiner og rekruttere andre immunceller. Dette forsterker den inflammatoriske responsen.
- Induksjon av trombose: NETs kan aktivere koagulasjonskaskaden og bidra til tromboser.
Implikasjoner av NETs for revmatiske sykdommer: NETs er implisert i patogenesen av flere revmatiske sykdommer:
- ANCA-vaskulitt: Autoantistoffer mot MPO og PR3 som finnes i NETs, er karakteristiske for denne gruppen av vaskulitter.
- Systemisk lupus erythematosus (SLE): NETs bidrar til inflammasjon og organskade ved SLE.
- Revmatoid artritt (RA): NETs er funnet i synovialvæsken i leddene hos RA-pasienter og bidrar til ledd-destruksjon.
- Psoriasis: NETs er involvert i hudinflammasjonen ved psoriasis.
-Ekstracellulære vesikler (EV)
Ekstracellulære vesikler (EV) er membranbundne partikler som frigjøres fra celler og fungerer som signalstoffer for intercellulær kommunikasjon. Vesiklene kan transportere et biologiske molekyler som proteiner, lipider, DNA, mRNA og mikroRNA (miRNA). EV klassifiseres hovedsakelig etter størrelse og biogenese i tre hovedtyper:
- Eksosomer: Små vesikler (30-150 nm) som dannes ved invaginasjon av endosomer og frigjøres ved fusjon med cellemembranen.
- Mikrovesikler (eller mikropartikler): Større vesikler (100-1000 nm) som avspaltes direkte fra cellemembranen.
- Apoptotiske legemer: Større fragmenter (500-5000 nm) som frigjøres fra celler under apoptose.
EV spiller en viktig rolle i en rekke fysiologiske og patologiske prosesser:
- Intercellulær kommunikasjon: EV overfører biologiske molekyler mellom celler og påvirker mottakercellenes funksjon.
- Immunregulering: EV kan modulere immunresponser ved å presentere antigener, overføre cytokiner og påvirke aktiveringen av immunceller.
- Inflammasjon: EV kan bidra til inflammasjon ved å transportere pro-inflammatoriske mediatorer.
- Koagulasjon: EV kan aktivere koagulasjonskaskaden.
EV er funnet i økte nivåer i biologiske væsker (synovialvæske og serum) hos pasienter med flere revmatiske sykdommer: Sjøgrens sykdom: EV er involvert i inflammasjonen i spyttkjertlene . ): EV bidrar til inflammasjon og ledd-destruksjon (Zhang B, 2021). : EV kan spille en rolle i nedbrytningen av brusk (Yang B, 2023).
EV fra immunceller som NK-celler, makrofager, monocytter og dendrittiske celler, bidrar til patogenesen av revmatiske sykdommer.
EV har et terapeutisk potensiale. De kan modifiseres for å frakte medikamenter eller terapeutiske molekyler direkte til målceller. Det kan forbedre effekten av behandlingen og redusere bivirkninger
-Prostaglandiner
Prostaglandiner (PG) er en gruppe lipidmediatorer som tilhører eikosanoidfamilien. De syntetiseres fra arakidonsyre og spiller en sammensatt rolle i fysiologiske og patologiske prosesser inkludert inflammasjon, smerte, feber, blodkoagulasjon og beskyttelse av mage-tarmslimhinnen. PG virker lokalt (autokrint og parakrint) og har ofte flerfoldige effekter i ulike vev (Wojcik P, 2021).
Syntesen av prostaglandiner starter med frigjøring av arakidonsyre fra cellemembranens fosfolipider ved hjelp av enzymet fosfolipase A2 (PLA2). Arakidonsyre omdannes deretter til prostanoider (inkludert prostaglandiner, tromboxaner og prostacyklin) av enzymet cyklooksygenase (COX). Det finnes to hovedisoformer av COX:
- COX-1 (konstitutiv COX): Uttrykkes i de fleste vev og er involvert i produksjonen av prostaglandiner som er viktige for normale fysiologiske funksjoner som beskyttelse av mage-tarmslimhinnen, regulering av nyrefunksjon og blodplateaggregering.
- COX-2 (induserbar COX): Uttrykkes lavgradig i de fleste vev under normale forhold, men induseres kraftig av proinflammatoriske cytokiner (f. eks. IL-1β, TNF-α), vekstfaktorer og endotoksiner. COX-2 er hovedenzymet som er ansvarlig for produksjonen av PG under inflammasjon.
PG metaboliseres raskt av enzymer som 15-hydroksyprostaglandin dehydrogenase (15-PGDH) som begrenser den lokale virkningen.
Virkninger:
Ulike PG har forskjellige og noen ganger motstridende effekter:
- Prostaglandin E2 (PGE2):
- Inflammasjon: Øker inflammasjon ved å øke vaskulær permeabilitet, rekruttere immunceller og sensibilisere nerveender for smerte.
- Feber: PGE2 er en viktig mediator for feber. Det virker på det preoptiske området i hypothalamus (temperatursentrum) og øker kroppstemperaturen. Pyrogener som IL-1β, IL-6 og TNF-α (endogene pyrogener) eller lipopolysakkarider (LPS) fra bakterier (eksogene pyrogener) induserer produksjonen av PGE2.
- Smerte: PGE2 sensibiliserer nerveender for smertefulle stimuli og bidrar til hyperalgesi (økt smertefølsomhet).
- Prostaglandin D2 (PGD2) er involvert i allergiske reaksjoner, søvnregulering og inflammasjon.
- Prostacyklin (PGI2):
- Vasodilatasjon: Dilaterer blodårer og hemmer blodplateaggregering.
- Beskyttelse av mage-tarmslimhinnen: Beskytter mage-tarmslimhinnen ved å øke produksjonen av mucus og bikarbonat.
- Tromboxan A2 (TXA2): Produseres hovedsakelig i blodplater og fremmer blodplateaggregering og vasokonstriksjon.
Prostaglandiner, spesielt PGE2, spiller en sentral rolle i patogenesen av flere revmatiske sykdommer:
- Revmatoid artritt (RA): PGE2 bidrar til inflammasjon, smerte og ledd-destruksjon.
- Artrose: PGE2 er involvert i nedbrytningen av brusk og smerte ved artrose.
- Andre inflammatoriske leddsykdommer: Prostaglandiner bidrar til inflammasjon og smerte ved en rekke andre inflammatoriske leddsykdommer.
Ikke-steroide antiinflammatoriske midler (NSAIDs) hemmer enzymet COX og dermed produksjonen av prostaglandiner.
- Tradisjonelle NSAIDs: Hemmer både COX-1 og COX-2. Hemming av COX-1 kan føre til gastrointestinale bivirkninger (magesår, blødninger) på grunn av redusert beskyttelse av mage-tarmslimhinnen.
- Selektive COX-2-hemmere (koksiber): Hemmer hovedsakelig COX-2 og har derfor en lavere risiko for gastrointestinale bivirkninger sammenlignet med tradisjonelle NSAIDs. Imidlertid har bruk av COX-2-hemmere vært assosiert med økt risiko for kardiovaskulære hendelser hos enkelte pasientgrupper.
Prostaglandiner samvirker med andre inflammatoriske mediatorer som cytokiner (f. eks. IL-1β, TNF-α) og leukotriener i komplekse nettverk som regulerer inflammasjon og smerte.
-Granulomer
Granulomer er knuter i som dannes i vevet ved en langvarig immunrespons. De består av en organiserte spesialiserte immunceller og er kroppens måte å begrense/kapsle inn en infeksjon eller et autoantigen som immunsystemet ikke klarer å eliminere raskt.
Kjernen i et granulom er tettpakket av makrofager. Disse kan omdannes til store epiteloidceller. Flere epiteloidceller kan smelte sammen og danner da store, flerkjernede kjempeceller (Langhans-kjempeceller). Hele strukturen er omgitt av en krans av lymfocytter (Pagan AJ, 2018).
- Granulomatose med polyangiitt (GPA/Wegeners granulomatose). Granulomer finnes i øvre og nedre luftveier samt i nyrene. Immunforsvaret angriper her feilaktig blodårene (vaskulitt), noe som utløser granulomdannelsen.
- EGPA (Churg-Strauss vaskulitt) er beslektet med GPA.
- Sarkoidose er ikke en klassisk revmatisk sykdom, men revmatologer er likevel ofte involvert i diagnostiseringen og behandlingen. Sykdommen er definert ved ikke-nekrotiserende granulomer i ulike organer som lunger, hud, og lymfeknuter.
- Revmatoid artritt (RA) revmatoid noduli (revmaknuter) består av granulomer (Parperis K, 2023).
- Granulomer ved andre sykdommer omfatter granuloma annulare (selvbegrensende, plakk og papler, pseudo-revmanodulus), necrobiosis lipoidica (på legger ved diabetes mellitus), mykobakterielle granulomer (tbc, atypiske mykobakterier), fremmedlegeme-granulom, nekrobiotisk xanthogranulom, sopp, syfilis, berylliose (Wiliams O, 2022). Generelt ses granulomer hyppigst hos pasienter med immundefekter (van Stigt AC, 2024).
4. Klinisk relevans: diagnostikk og behandling
Hvordan brukes denne immunologiske kunnskapen i klinisk praksis hos revmatologer? Her følger prinsipper og eksempler.
Diagnostiske og prognostiske implikasjoner
-
Autoantistoffer: RF, anti-CCP, ANA, anti-dsDNA, anti-Ro/La, anti-SCL-70 osv. kan hjelpe for diagnose, prognose, og kategoriinndeling av sykdom.
-
Biomarkører for inflammasjon: CRP, SR, cytokinnivåer (i enkelte tilfeller) – sier noe om aktivitet, respons på behandling.
-
Genetisk testing: HLA-alleler (f.eks. HLA-DRB1 i RA, HLA-B27 i spondyloartritter), polymorfismer i immunsignalgener kan gi risiko.
-
Biopsi/histologi: Synovialbiopsi (inflammasjon, lymfocytter/lymfefollikler, neovaskularisering), nyrebiopsi i SLE, osv.
Behandling: immunmodulering og immundempende legemidler (DMARDs)
Overordnede mål er å kontrollere inflammasjon, hindre vevsskade og funksjonstap, oppnå remisjon eller lav sykdomsaktivitet og minimere bivirkninger, spesielt infeksjoner.
Klassiske immunosuppressive og sykdomsmodifiserende midler (csDMARDs) er metotrekat: basis-behandling ved mange revmatiske sykdommer (RA, psoriasisartritt), sulfasalazin, leflunomid, azatioprin og mykofenolatmofetil.
Kortikosteroider i ved behov for rask inflammasjonsdemper
Biologiske legemidler (bDMARDs)
-
TNF-hemmere: mange RA-pasienter responderer godt; også ved annen inflammatorisk artritt.
-
IL-6 hemmere: f.eks. tocilizumab – nyttig spesielt i sykdommer hvor IL-6 dominerer
-
IL-1 hemmere i enkelte tilstander (f.eks. autoinflammatoriske syndromer)
-
IL-17 / IL-23 akser i spondyloartritter og psoriasisartritt
-
B-celle depletering: f.eks. rituximab, relevant i RA, vaskulitt og SLE
-
T-celle kostimulatoriske blokkader: CTLA-4 agonister eller CD28 antagonister
JAK-hemmere (tsDMARDs): små molekyler som hemmer intracellulære signalveier av flere cytokiner.
CAR T-cellebehandling og BitEs. Med økende kjennskap til immunsystemets funksjoner har vi oppnådd medikamenter som kontrollerer inflammatoriske revmatiske sykdommer effektiv på en måte vi ikke har sett maken til tidligere. Enda mer kunnskap på dette område kombinert med genteknologiske fremskritt antas å kunne forbedre behandlingsmulighetene ytterligere.
Spesielle hensyn ved immunsupprimerende behandling
-
Risiko for infeksjon: screening (tuberkulose, hepatitis), vaksinasjoner, monitorering.
-
Kant av immunmodulering og kreft: nøye vurdering ved langvarig immunsuppresjon.
-
Behandling tilpasset pasient (alder, komorbiditet, ønsket graviditet etc.).
Litteratur
Scherer HU, 2020 (etiologi ved revmatoid artritt)
Zimmermann KA, 2018 (Immune system, Lifescience)
Murphy, Travers & Wa/port. Janeway’s Immunobiology. Garland Science.
Teksten er skrevet og gjennomgått av forfatterne. I bearbeidelsen har vi brukt kunstig intelligens i noen avsnitt.

{kind=link}
{kind=link}