BARN MED REVMATISK SYKDOM. BARNEREVMATOLOGI (REV 053-062)
123 Juvenil Mixed Connective Tissue Disease (JMCTD). REV 021, REV 054
Øyvind Palm
Kjennetegn på MCTD
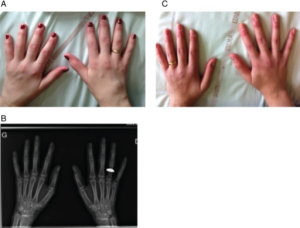
Raynauds fenomen, hovne (“puffy”) fingre er tidlige symptomer.
Overlappende symptomer med andre bindevevssykdommer som eksantem (SLE), sklerodaktyli (systemisk sklerose) og lavgradig myositt med eksantem (juvenil dermatomyositt)
ANA er oftest positiv, og sub-gruppen RNP er obligatorisk.
Definisjon
Dersom Mixed Connective Tissue Disease (MCTD) er en variant av Mixed Connective Tissue Disease (MCTD) som debuterer før 18 års alder. Denne tidlige sykdomsstarten kan medføre et annet sykdomsforløp enn hos voksne. Alle pasienter med JMCTD har anti-RNP (ribonukleoprotein) antistoffer (Badu ED, 2023).
Forekomst
JMCTD er en sjelden tilstand, med en årlig insidens på 0,8-5 tilfeller per million barn (Pelkonen PM, 1994). Juvenil debut observeres hos 7-23 % av alle med MCTD (Kotajima L, 1996; Burdt MA, 1999).
Symptomer
De første sykdomstrekk ved JMCTD er vanligvis Raynauds fenomen og hovne (puffy») hender. Ved høy inflammatorisk aktivitet kan allmenntilstanden være påvirket med feber, utmattelse og hovne lymfeknuter.
En omfattende litteraturgjennomgang viste følgende fordeling av symptomene (Terminello A, 2024):
- Raynauds fenomen (69.7%)
- Artritt (60.9%)
- Muskelpåvirkning (53.5%)
- Hudmanifestasjoner (39.5%)
- “Puffy fingers/ hands” (29.3%)
- Artralgi (25.6%), feber (22.3%)
- Lungemanifestasjon (14.4%)
- Sklerodaktyli (13.5%)
- Lymfadenopati (10.7%)
- Serositt (10.2%)
- Øsofagusmanifestasjon (6.9%),
- Nervesystemmanifestasjon (6.9%)
- Tørre øyne (3.7%)
- Tørr munn (3.7%)
- Hepatosplenomegali (2.8%)
- Kardial manifestasjon(2.8%)
- Hepatitt (2.3%)
- Parotitt (betennelse i spyttkjertler) (2.3%)
- Hashimotos thyreoiditt (0.9%)
- Øyemanifestasjon (0.9%)
Undergrupper av JMCTD
Man kan dele symptomene inn i tre hovedgrupper:
- SLE-lignende type. Dette er den vanligste formen ved debut og kjennetegnes ved artritt, ansikts-erythem og feber. Blodprøver viser ofte leukopeni og/eller trombocytopeni
- Systemisk sklerose-lignende type kjennetegnes ved sklerose / fibrose i hud. I forløpet utvikler ca. 1/3 lungefibrose, noe som er uvanlig ved SLE-lignende type. Forekomst av pulmonal hypertensjon er noe økt i denne gruppen, men antakelig ikke høyere enn 3-4% (Gunnarsson R 2013).
- Polymyositt/dermatomyositt-lignende type er den mest sjeldne formen. Typisk er myositt, artritt og serositt (pleuritt og perikarditt)
I sykdomsforløpet endrer symptomene av de tre hovedgruppene seg noe. SLE- og myositt lignende typer får mindre fremtrede symptomer, mens systemisk sklerose-typen har progredierende symptomer.
Undersøkelser
Anamnesen bør dekke de mest aktuelle symptomer (se ovenfor). Spør spesielt etter tegn på Raynauds fenomen, hudforandringer, lungesymptomer og tegn til redusert muskelstyrke.
Klinisk undersøkelse: Vurder hjerte, lunger, blodtrykk, hår, hud, ledd og muskulatur.
Laboratorieprøver kan omfatte CRP, SR, Hb, leukocytter med differensialtellinger, elektrolytter, IgG, glukose, lever-, nyre- og thyreoidea-funksjonsprøver, CK, albumin, samt antistoff (se mer nedenfor) ANA, anti-DNA, komplement C3, C4 og urin stiks.
- Høy CRP ses ved serositt (pleuritt, perikarditt), uttalt artritt og kompliserende infeksjoner.
- Cytopenier påvises ofte ved SLE-lignende type. Mild leukopeni (og lymfopeni) er vanligst.
- Forhøyet CK indikerer myositt.
- Urin-undersøkelser er viktige for å utelukke tegn på aktiv nefritt, men dette er sjelden. Urinen undersøkes for proteiner, erytrocytter og ved utslag gjøres mikroskopi (sylindre) og protein/kreatinin ratio estimeres.
Immunologiske undersøkelser. ANA forventes å være positiv hos nesten alle og anti-RNP subgruppen er obligatorisk (pr. definisjon). Antifosfolipid antistoffer (lupus antikoagulant, kardiolipin og beta-2 glykoprotein antistoff) forekommer hos enkelte og disponerer da for tromboembolier / antifosfolipid syndrom.
Bildediagnostikk
- Ultralyd eller MR av ledd kan gjøres for å verifisere artritt eller annen synovitt.
- CT av lunger bør gjøres for å vurdere om lungemanifestasjoner foreligger.
- EKG kan utelukke tegn til arytmi.
- Ved systemisk sklerose-form er ekkokardiografi aktuelt for å utelukke (sjeldne) tegn til pulmonal hypertensjon (PAH).
Komorbiditet
Det er økt forekomst av andre autoimmune sykdommer som: hypothyreose (Hashimoto), autoimmun hepatitt, autoimmun pankreatitt og makrofag aktiveringssyndrom (MAS/HLAH) (Parvaneh JC, 2019). Kronisk sykdom øker forekomsten av psykiatriske tilstander, inklusiv alvorlig depresjon, psykose.
Klassifikasjonskriterier
Det finnes ikke separate kriterier for juvenil MCTD. Man bruker kriteriene for MCTD som for voksne (Kasukawa-kriteriene, Alarcon-Segovia er mest brukt)
Retningslinjer
Nasjonale anbefalinger (NAKBUR)
Differensialdiagnoser
Differensialdiagnoser for juvenil MCTD (jMCTD) som opptrer i barnealder kan være utfordrende, da sykdommen har overlappende symptomer med andre bindevevssykdommer. Her er noen viktige differensialdiagnoser å vurdere:
- Juvenil Systemisk lupus erythematosus (jSLE): SLE kan ligne jMCTD med symptomer som leddsmerter, utslett, tretthet og feber. SLE har ofte mer spesifikke symptomer som sommerfuglutslett i ansiktet og nyreaffeksjon, samt karakteristiske autoantistoffer (som anti-dsDNA) som ikke alltid er tilstede ved jMCTD.
- Juvenil dermatomyositt (JDM): JDM kan ligne jMCTD ved å forårsake muskelsvakhet, hudutslett og lett forhøyede muskelenzymer i blodet. JDM har ofte mer uttalte muskelsymptomer og karakteristiske hudutslett som Gottrons papler og heliotropt utslett.
- Juvenil idiopatisk artritt (JIA): JIA kan ligne jMCTD med leddbetennelse, men mangler vanligvis de systemiske symptomene og autoantistoffene som er typiske for jMCTD.
- Juvenil Systemisk sklerose: Systemisk sklerose kan ligne jMCTD ved å forårsake Raynauds fenomen og hudfortykkelse, men har ofte mer uttalte fibrotiske forandringer i huden og indre organer.
Behandling
Behandlingen er tverrfaglig og involverer leger fra ulike spesialiteter, fysio- og ergoterapeuter.
- NSAIDs. Brukes mot smerter, spesielt leddsmerter.
- Kortikosteroider (Prednisolon) brukes ved behov i korte perioder ved myositt, alvorlig serositt eller uttalt artritt.
- csDMARDs. Hydroksyklorokin (Plaquenil) kan muligens beskytte mot komplikasjoner, men god dokumentasjon mangler. Metotreksat er mest brukt mot artritt eller myositt. Azathioprin (Imurel) virker mot cytopenier, serositt, vaskulitt og interstitiell lungesykdom. Mykofenolat kan brukes ved interstitiell lungesykdom.
- Biologiske legemidler er sjelden indisert.
Prognose
Systemisk sklerose-lignende type har generelt dårligere prognose enn SLE- og myositt-formene. Likevel er dødeligheten samlet sett ikke funnet å være signifikant økt sammenlignet med normalbefolkningen. En norsk studie fant en mortalitetsrate på 5,1% over en periode på 16,8 år (3 per 1000 pasienter) (Hetlevik SO, 2017).
Litteratur
