ANDRE SYKDOMMER (REV 063-REV 077)
181 Marfans syndrom (REV 063)
Øyvind Palm
Kjennetegn på Marfans syndrom
Påfallende stor kroppshøyde, skoliose, lange, armer og ben (dolichostemomeli) med lange fingre og tær (arachnodaktyli) i forhold til kroppens lengde.
Brystben deformiteter som fuglebryst (pectus carniatum).
Hypermobile ledd, linse-subluksasjon, risiko for aortaaneurismer
- Karpaltunnelsyndrom, Bakers cyste, entesopati, lumbago, isjas, peritendinitt og kapsulitt i skulder er omtalt i egne kapitler.
ICD-10: Q87.4
Definisjon
. CC BY-2.0")
Marfans syndrom er en av de vanligste genetiske sykdommene som påvirker muskel- og skjelettsystemet. Sykdommen kan imidlertid også ha alvorlige konsekvenser for andre organer, spesielt hjertet og blodårene i form av aneurismer og disseksjoner. Symptomer og alvorlighetsgrad varierer betydelig, fra isolerte manifestasjoner til multiorganaffeksjon. Marfans syndrom er en monogenetisk sykdom, noe som betyr at den skyldes en mutasjon i ett enkelt gen. Den skiller seg dermed fra de systemiske autoimmune bindevevssykdommene (Zeigler S, 2021).
Sammen med en vaskulær Ehlers-Danlos syndrom, Loeys-Dietz syndrom og hereditært (arvelig) thorakalt aortaaneurisme (HTAD) utgjør Marfans syndrom gruppen av arvelige “Marfan-lignende” sykdommer (Asta L, 2023).
Marfans syndrom utredes ofte innen fysikalsk medisin. Revmatologer spiller også en viktig rolle i å skille Marfans syndrom fra andre tilstander, henvise pasienter til riktig spesialist og bidra til tverrfaglig samarbeid for å sikre optimal behandling og oppfølging av disse pasientene.
Ved etablert diagnose kan også TRS kompetansesenter for sjeldne diagnoser ved Sunnaas sykehus HF bidra med informasjon om håndtering og pasientinformasjon.
Genetikk og patogense
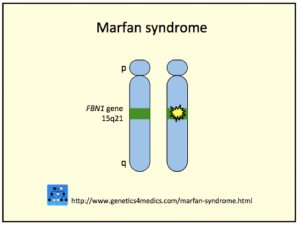
Marfans syndrom skyldes nesten alltid en mutasjon i genet FBN-1-genet på kromosom 15 som koder for proteinet fibrillin-1 (Hilhorst-Hofste Y, 2010). Fibrillin-1 er et matriks glykoprotein og hovedbestanddelen i elastiske fibre i bindevevet. I mindre enn 10% av tilfellene finner man ikke mutasjon i FBN1 genet (Salik I, 2023).
En kombinasjon av strukturelle forandringer i mikrofibre, forhøyet TGF-beta og overekspresjon av matriksmetalloproteinasene MMP-2 og MMP-9 medfører forandringer i vevet ved Marfans syndrom (Hoffjan S, 2012). Histologisk kan man i aortaroten påvise cystisk medianekrose, fibrose og tap av glatt muskulatur. Fragmentering av elastin er ofte mer uttalt enn hos personer uten bindevevssykdom (Collins MJ, 2008).
Epidemiologi
Insidensen av Marfans syndrom estimeres til 1-3 per 10 000 (Gray JR, 1994). Alle etnisiteter rammes, og kvinner og menn er like ofte påvirket. Selv om Marfans sykdom er blant de vanligste genetiske sykdommene, defineres den som en sjelden sykdom (færre enn 1 pr 2.000 personer i henhold til Det Norske Helse og Omsorgs-departementet).
Symptomer og manifestasjoner
- Skjelettsystemet: Påfallende stor kroppshøyde (Erkula G, 2002). Skoliose. Lange, armer og ben (dolichostemomeli) med lange fingre og tær (arachnodaktyli) i forhold til kroppens lengde. Pes planus (plattfot) skyldes hyperelastiske ligamenter (Lindsey J, 1998). Brystben deformiteter som fuglebryst (pectus carniatum). Overbevegelige (hypermobile) ledd (Kaissi AA, 2013).
- Øyne: Linse subluksasjon (ektoptisk linse, ectopia lentis) påvises med spaltlampe (øyelege) hos 50-80% og er vanligste øye-manifestasjon. Samtidig er utgjør Marfans syndrom de fleste tilfeller av påvist ektoptisk linse. Ofte foreligger også synsforstyrrelser med myopi, tidlig katarakt og astigmatisme (Zech J-C, 2020). Pasienten undersøkes derfor av øyelege regelmessig.
- Hjertet og blodårer: Dette er de alvorlige komplikasjonene. Aortarot sykdom med insuffisiens i aortaklaffen, aneurisme med dilatasjon og disseksjon er den mest alvorlige komplikasjonen ved Marfans syndrom med en estimert mortalitet på opp til 60-80% (Salik I, 2021). Spesiell oppmerksomhet i svangerskap er viktig. Mitralklaff prolaps ses hos 40% -54% (Rybczynski M, 2010). Også rytmeforstyrrelser er vanlig.
- Lunger: Flere typer lungemanifestasjoner forekommer. Spontan pneumothoraks er sjelden, men relativt hyppig blant de yngre pasientene. Andre manifestasjoner er kronisk bronkitt, emfysem og bronkiektasier (Zarfati A, 2023).
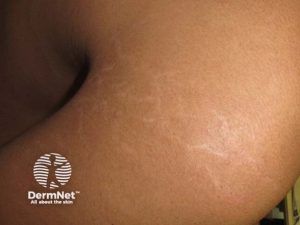
- Hud. Striae skyldes hypermobilt bindevev.
Diagnosen
Diagnosen Marfans syndrom stilles basert på kliniske kriterier kriterier for diagnosesetting (Ghent-kriteriene fra 2010) (2010 Ghent), familiehistorie og genetiske tester (Loeys BL, 2010):
Uten arvelig disposisjon:
- Aortarot utvidet (Z-skår ≥2) og ektopitisk linse
- Aortarot utvidet (Z-skår ≥2) og FBN1 mutasjon (gentest)
- Aortarot utvidet (Z-skår ≥2) og systemisk skåring* >7 punkter (spesielt skåringssystem*)
- Ektopisk linse og FBN1 mutasjon med kjent aortaaffeksjon
Med familie med Marfans sykdom:
- Ektoptisk linse
- Systemisk skåring* Aortarot utvidet ≥7
- Aortarot utvidet (Z-skår ≥2)
*Skår: Pectus carniatum=2 (pes excavatum eller asymmetri=1), Malleol-deformitet (ankel)=2 (plattfot=1), Dural ektasi=2, Protrusio acetabuli=2, Dys-proporsjonal armlengde =1, Skoliose eller kyfose=1, Redusert albue ekstensjon=1, Ansiktsskjelett forandringer= 1, Striae i huden=1, myopi=1, Mitralprolaps =1.
Gamle kriterier: major kriterier (minst 4):
- Redusert proporsjon overkropp/underkropp ratio (0,85 vs 0,93 hos normale)
- Arachnodactyli (fingre, tær), positiv tommel-håndledd-test)
- Skoliose mer enn 20 grader eller spondylolistese
- Medial malleol-feilstilling som gir pes planus (plattfot)
- Redusert ekstensjon i albuer (<170 grader)
- Pectum carniatum (fuglebryst) (Differensialdiagnoser: Morquio syndrome, Noonan syndrome, Trisomi 18, Trisomi 21, Homocystinuri, Osteogenesis imperfecta, multiple lentigines syndrom, and Sly syndrome)
- Pectus excavatum (traktbryst) som krever kirurgi
- Protrusio Acetabuli (Differensial diagnoser: Paget syndrom, revmatoid artritt, Bekhterevs sykdom, Osteomalasi)
Genetikk: Prøve kan sendes Oslo Universitetssykehus, Ullevål. Mer om genetisk test her (genetikkportalen.no).
Differensialdiagnoser
Marfans syndrom kan i likhet med en del andre tilstander føre til en rekke helseproblemer, inkludert hjerteproblemer, øyeproblemer og skjelettabnormaliteter.
- Ehlers-Danlos syndrom: En gruppe arvelige tilstander som kjennetegnes av hypermobilitet i ledd, elastisk hud og skjørhet i vev som kan ligne Marfan syndrom ved tilstedeværelse av skjelett- og hjerteproblemer.
- Ektoptisk linse syndrom (ELS): En sjelden tilstand med løs eller dislokert linse i øyet, som kan ligne Marfan syndrom der linseluksasjon også er en vanlig funn.
- Familiær thorakalt aorta aneurisme og disseksjon syndrom (FTAAD/FTAA): n arvelig tilstand som øker risikoen for utvidelse og disseksjon av aorta, som kan ligne Marfan syndrom ved tilstedeværelse av hjerte-kar-problemer.
- Hereditært (arvelig) thorakalt aortaaneurisme (HTAD): En tilstand som kjennetegnes av utvidelse av aorta i brystkassen som kan ligne Marfan syndrom ved tilstedeværelse av hjerte-kar-problemer.
- Homocystinuri: En stoffskiftesykdom som kan gi symptomer som ligner på Marfan syndrom, inkludert lange lemmer, arachnodactyly (lange fingre og tær) og linseluksasjon.
- Hypermobilitetssyndromet: En tilstand preget av økt leddbevegelighet uten andre underliggende systemiske funn som kan forveksles med Marfan syndrom på grunn av felles leddmanifestasjoner.
- Klinefelter syndrom: En tilstand som rammer menn og er karakterisert av ekstra X-kromosom som i noen tilfeller kan gi høyere kroppsbygning som kan overlappe med Marfan syndrom.
- Kongenital kontraktær araknodaktyli (CCA): En sjelden arvelig tilstand som kjennetegnes av medfødte kontrakturer i ledd og arachnodactyly som kan ligne Marfan syndrom ved tilstedeværelse av skjelettproblemer.
- Loeys-Dietz syndrom: En arvelig bindevevssykdom som ligner på Marfan syndrom, men som ofte har mer alvorlige hjerte-kar-manifestasjoner og karakteristiske ansiktstrekk.
- Menkels sykdom (defekt i kobber-metabolisme): En sjelden arvelig tilstand som påvirker kobbermetabolismen som kan gi symptomer som ligner på Marfan syndrom, inkludert skjelett- og hjerteproblemer.
- Noonan syndrom: En genetisk tilstand som kan gi kortvoksthet, hjerteproblemer og karakteristiske ansiktstrekk, som i noen tilfeller kan overlappe med Marfan syndrom.
- Pseudoxanthoma elasticum: En sjelden arvelig tilstand som påvirker bindevevet i hud, øyne og blodårer som kan ligne Marfan syndrom ved tilstedeværelse av hud- og øyeforandringer.
- Shprintzen-Goldberg syndrom (SGS): En sjelden arvelig tilstand som kjennetegnes av skjelett-, hjerte- og nevrologiske problemer som kan ligne Marfan syndrom ved tilstedeværelse av skjelett- og hjerteproblemer.
- Stickler syndrom (hereditær arthro-oftalmopati): En gruppe arvelige tilstander som påvirker bindevevet, spesielt i øyne, ledd og ører, som kan ligne Marfan syndrom ved tilstedeværelse av øye- og leddproblemer.
- Turner syndrom: Rammer kvinner og er karakterisert av manglende eller skadet X-kromosom, som i noen tilfeller kan gi kortvoksthet og hjerteproblemer, som kan overlappe med Marfan syndrom.
- Weill-Marchesani syndrom (WMS): En sjelden arvelig tilstand som kjennetegnes av kortvoksthet, stive ledd og øyeproblemer som kan ligne Marfan syndrom ved tilstedeværelse av øye- og leddproblemer.
Behandling
Behandlingen av Marfans syndrom er rettet mot å forebygge og behandle komplikasjoner relatert til bindevevssvakhet, spesielt i hjerte, skjelett og øyne. Det finnes ingen kurativ behandling og behandlingsstrategien fokuserer på symptomlindring, livsstilsendringer og individualiseres basert på pasientens spesifikke symptomer og alvorlighetsgrad av sykdommen.
Tverrfaglig samarbeid mellom kardiologer, ortopeder, øyeleger og genetikere er essensielt.
Kardiovaskulær behandling:
- Betablokkere:
- Disse medikamentene reduserer hjertefrekvensen og blodtrykket, og dermed belastningen på aorta.
- De bidrar til å forebygge aortadisseksjon og -ruptur.
- Angiotensin II-reseptorantagonister (ARB): Kan brukes som et alternativ eller tillegg til betablokkere for å redusere belastningen på aorta.
- Aortakirurgi.
- Nødvendig ved betydelig utvidelse av aorta (aortaaneurisme) for å forebygge ruptur eller disseksjon.
- Aortarot- eller ascendensutskifting utføres når aortadiameteren når en kritisk størrelse. Karkirurger / thorakskirurger har retningslinjer for når operasjoner bør gjøres. Gjennomsnittsalder for operasjon er 32 år.
- Hjerteklaff kirurgi. Mekaniske hjerteklaffer foretrekkes på grunn av lang holdbarhet. Pasientene trenger da livslang antikoagulasjonsbehandling. For pasienter som ikke kan bruke antikoagulantia, er klaffebevarende kirurgi aktuelt.
Skjelettbehandling:
- Ortopedisk oppfølging:
- Regelmessig vurdering av skjelettdeformiteter som skoliose, pectus deformitet og fotdeformiteter.
- Bruk av korsett eller ortoser for å korrigere skoliose.
- Kirurgisk korreksjon:
- Nødvendig ved alvorlige skjelettdeformiteter som påvirker funksjon eller forårsaker smerte.
- Fysioterapi:
- For å opprettholde bevegelighet, og styrke støttemuskulatur.
- Ortoser: Stabiliserende ortoser og tilpasset fottøy kan være nødvendlig.
Øyebehandling:
- Regelmessige øyeundersøkelser: Viktig for å oppdage og behandle linseluksasjon, netthinneløsning og andre øyeproblemer.
- Briller eller kontaktlinser: For å korrigere synsfeil.
- Kirurgi: Nødvendig ved linseluksasjon eller netthinneløsning.
Livsstilsråd:
- Unngåelse av anstrengende aktiviteter: For å redusere risikoen for aortadisseksjon eller -ruptur.
- Regelmessig oppfølging: Viktig for å overvåke sykdomsforløpet og justere behandlingen etter behov.
- Genetisk veiledning: Viktig, da dette er en arvelig sykdom.
Kontroller: Regelmessig oppfølging med bildediagnostikk (MR-angiografi, ultralyd Doppler/ekkokardiografi, CT) er viktig for å overvåke aorta og andre blodårer.
Merk: Kortikosteroider og annen immundempende behandling har ingen plass i behandlingen av genetiske bindevevssykdommer, til forskjell fra autoimmune bindevevssykdommer.
Prognose
Det eksisterer per dags dato ingen kurativ terapi for Marfans syndrom. Imidlertid har prognosen gjennomgått betydelig forbedring i løpet av de siste tiårene. Dette illustreres ved en økning i median forventet levealder fra 48 år i 1972 til 72 år i 1993 (Silverman DI, 1995).
Det må antas at prognosen har blitt ytterligere forbedret i ettertid. Dette tilskrives effekten av regelmessig medisinsk oppfølging, som inkluderer konsultasjoner hos kardiolog, vaskulær billeddiagnostikk for deteksjon og oppfølging av aneurismer, samt behandling av pneumotoraks. Menn har generelt en redusert forventet levealder sammenlignet med kvinner
Litteratur
Kompetansesenter, Sunnås Sykehus
