ANDRE SYKDOMMER (REV 063-REV 077)
214 Schnitzlers syndrom (REV 076, REV 080, REV 189)
Øyvind Palm and Jan Tore Gran
Kjennetegn på Schnitzlers syndrom
Sjelden autoinflammatorisk sykdom med kronisk kløende eksem, skjelettsmerter og residiverende feber med intermitterende topper på over 40 °C hos 90 %.
Monoklonal M-komponent i serum-elektroforese.
- Autoinflammatoriske sykdommer, hudmanifestasjoner og blodsykdommer er også beskrevet i egne kapitler.
ICD-10: L50.8
Definisjon

Schnitzlers syndrom er en sjelden, ervervet autoinflammatorisk sykdom karakterisert ved et urtikarielt, ikke-kløende utslett og tilstedeværelse av en M-komponent (monoklonalt protein i form av IgM-κ, sjelden IgG) paraprotein i serum-elektroforese). Sykdommen debuterer nesten alltid etter 40-årsalderen (Simon A, 2013).
Historie
Sykdommen ble først beskrevet av den franske hudlegen Liliane Schnitzler i 1972 (L. Schnitzler, Lésions urticariennes chroniques permanentes (érythème pétaloïde?) Cas cliniques No 46 B, J Dermatol Angers, 1972).
Forekomst
Schnitzlers syndrom oppstår nesten alltid etter 40 års alder, med en gjennomsnittlig debutalder rundt 50 år. Det er en liten overvekt av menn som rammes (Simon A, 2013).
Patogenese
Årsaken (etiologien) til Schnitzlers syndrom er ukjent, og det er ikke påvist bakenforliggende gen-mutasjon. I sykdomsforløpet sees økt sekresjon av interleukin-1β (IL-1β) og IL-6, noe som er typisk for autoinflammatoriske sykdommer (Braud A, 2024). En mulig patogenetisk sammenheng med Waldenström makroglobulinemi er også foreslått.
Symptomer
- Allmenntilstanden er ofte redusert i form av utmattelse, vekttap og myalgi (Braud A, 2024).
. CC BY 2.0")
- Kronisk urticaria: Utslettet består av svakt røde papler (små, solide knuter) på 0,5-3 cm i størrelse, som varer i 12-24 timer. Nye lesjoner utvikles daglig og kan gi kløe sent i sykdomsforløpet. Utslettet er vanligvis lokalisert til overkroppen og ekstremitetene, og kan utløses av alkohol.
- Feber. Tilbakevendende, periodisk feber med intermitterende topper på over 40 °C forekommer hos 90 % av pasientene. Frysninger er sjeldne, og det er ingen sammenheng mellom feber og utslett.
- Skjelett-smerte forekommer hos 59 %, oftest i bekken og tibia. Dersom distale femur er affisert, kan kneet påvirkes, “hot knee”
- Lymfadenopati sees hos ca. 50% av tilfellene.
- Polynevropati (sjelden) kan være sensorisk og symmetrisk (Braud A, 2024).
| Symptomer og undersøkelsesfunn ved Schnitzlers syndrom (Simon A, 2013) | |
| Urtikarielt eksantem | 100% |
| Forhøyet SR (≥30) | 95% |
| Feber | 93% |
| Monoclonal IgM gammopati | 89% |
| Kappa lette kjeder | 89% |
| Artralgi/artritt | 77% |
| Leukocytose (≥10 000) | 76% |
| Skjelettsmerte | 68% |
| Abnormale Skjelettforandringer (radiologisk) | 62% |
| Palpable forstørrede lymfeknuter (lymfadenopati) | 47% |
| Pruritus | 45% |
| Lever- og/eller milt forstørrelse | 34% |
Undersøkelser
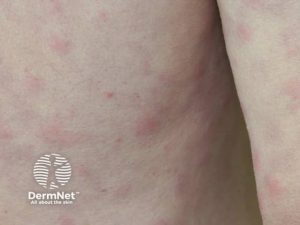
Diagnosen av Schnitzlers syndrom kan være vanskelig, da symptomene kan ligne på symptomer på andre tilstander (Puxkandi V, 2023).
Anamnesen kartlegger aktuelle symptomer (se ovenfor), særlig hos middelaldrende personer med uttalt urticaria og økt IgM i serum.
Klinisk er artritt og artralgi hyppig og urtikaria er obligatorisk. Feberepisoder, ledd- og skjelettsmerter, utmattelse, vekttap er vanlig (Gusdorf L, 2017). Hepatosplenomegali ses hos ca en av tre, lymfadenopati hos ca. 50 %. Polynevropati kan også forekomme.
Laboratorieprøver. Rutineprøver kan omfatte CRP, SR, celletellinger, elektrolytter, lever-, nyre- og thyreoidea-funksjonsprøver, glukose, CK, serumelektroforese og kvantitering av IgG, IgM og IgA. Urin stiks. Ved proteinuri kan også urin-elektroforese vurderes.
- Senkningsreaksjon (SR) er ofte forhøyet.
- Leukocytose hos 90 %.
- Serumelektroforese kan vise monoklonalt IgM, hvorav 89 % er assosiert med kappa-lette kjeder.
- Schnitzlers syndrom er en kronisk tilstand som kan utvikle seg til lymfoproliferativ lidelse (10 års risiko 15 %), oftest Waldenström makroglobulinemi, men dette kan to opp til 20 år. AA amyloidose rapportert.
Bildediagnostikk. Osteosklerotiske forandringer, ofte ved knær og i bekkenet hos ca. 40%. Røntgen- CT- eller MR. PET/CT-undersøkelser er mest sensitiv (Niederhauser BD, 2014) .
Biopsi. Histologisk sees neutrofil urticaria uten tegn til vaskulitt. I lymfeknuter kan reaktiv lymfadenitt sees.
Diagnose
Diagnosen kan bygge på Strasbourg diagnostiske kriterier:
Absolutte krav (major kriterier)
- Kronisk urticaria
- Monoklonal IgM eller IgG
Minor kriterier
- Residiverende feber (>38 grader, uten annen grunn)
- Objektive funn med skjelett-remodulering (ved PET, skjelettscintigrafi, MR eller benspesifikk alkalisk fosfatase) med eller uten skjelettsmerter
- Neutrofil infiltrasjon i huden ved biopsi (fravær av fibrinoid nekrose og vesentlig dermalt ødem)
- Leukocytose (neutrofile >10.000/mm3) og/eller økt CRP >30 mg/L
Sikker diagnose: Alle absolutte krav/major + IgM og totalt to minor kriterier, eller IgG paraprotein og totalt tre minor kriterier er oppfylt.
Sannsynlig diagnose: Absolutte krav + IgM inkludert ett minor kriterium, eller IgG inkludert to minor kriterier.
Differensialdiagnoser
- Autoinflammatoriske sykdommer inklusiv Muckle Wells syndrom, familiær middelhavsfeber og TRAPS syndrom.
- Juvenil artritt (systemisk type): Kan også gi feber, utslett og leddbetennelse, men rammer hovedsakelig barn og har et annet mønster av leddaffeksjon enn Schnitzlers syndrom.
- Kronisk idiopatisk urtikaria: Dette er en vanlig tilstand som kjennetegnes av elveblest som varer i mer enn seks uker. I motsetning til Schnitzlers syndrom er det ikke assosiert med feber eller monoklonal gammopati.
- Lymfom: Kan gi feber, elveblest og forstørrede lymfeknuter, som kan ligne på Schnitzlers syndrom.
- Malignitet: Andre former for kreft kan også gi feber og utslett, men har ofte andre symptomer og funn som kan hjelpe med å skille dem fra Schnitzlers syndrom.
- Stills sykdom i voksen alder adult Stills); ligner på systemisk juvenil artritt, men rammer voksne og kan også gi feber, utslett og leddbetennelse.
- Waldenstrøms sykdom: Dette er en annen type kreft som involverer lymfesystemet og kan gi lignende symptomer som Schnitzlers syndrom, inkludert monoklonal gammopati (IgM).
Behandling
Behandlingen av Schnitzler syndrom er rettet mot å lindre symptomer og forbedre livskvaliteten, da det ikke finnes noen kurativ terapi.
Biologisk behandling
- Interleukin-1 (IL-1)-hemmere: (anakinra, ev. canakinumab) er førstevalget basert på flere studier og rapporter og har vist seg svært effektivt i å kontrollere symptomene (Néel A, 2014 ; Kuemmerle-Deschner JB, 2016 ; Krause K, 2017).
- IL-6-hemmer: Tocilizumab (RoActemra) har vist effekt hos pasienter som ikke har hatt tilstrekkelig respons på IL-1-hemmere (Krause K, 2012). Ved manglende behandlingseffekt bør diagnosen revurderes.
Antihistaminer kan brukes for å lindre kløe og urtikaria, men er ofte ikke tilstrekkelig for å kontrollere alle symptomer.
Kortikosteroider. I sjeldne tilfeller kan kortikosteroider brukes for å redusere inflammasjon, men langvarig bruk er vanligvis ikke anbefalt på grunn av bivirkninger.
Ibrutinib har også vist seg å ha effekt ved å hemme tyrosinkinase.
Annet: Smertelindring med analgetika ved behov. Behandling av eventuelle komplikasjoner, som artritt eller feber.
Oppfølging: Regelmessig oppfølging er viktig for å overvåke sykdomsaktiviteten og eventuelle komplikasjoner, spesielt med tanke på utvikling av blodsykdommer som monoklonal gammopati av ukjent betydning (MGUS) eller lymfoproliferative sykdommer. Blodprøver og eventuelt benmargsundersøkelser kan være nødvendig.
Prognose
Schnitzler syndrom er en kronisk tilstand, men med moderne behandling er prognosen generelt god. Sykdomsaktiviteten kan følges med serum IgM og IgG. Omtrent 15-20% vil utvikle en lymfoproliferativ sykdom, oftest Waldenstöms makroglobulinemi (Simon A, 2013). AA amyloidose er også sett i forløpet (Gusdorf L, 2007). Livslengden er vanligvis likevel ikke vesentlig forkortet (de Koning HD, 2007).
Litteratur
Simonsen HE, 2022 (case)
Betrain A, 2020 (canakinumab)
Kacar M, 2019 (relasjon til autoinflammatorisk sykdom)
Gran JT, Midtvedt Ø, 2011 (case)
Gran JT, Midtvedt Ø, 2011 (behandling med anakinra)
