ANDRE SYKDOMMER (REV 063-REV 077)
187 Myopatier (non-inflammatoriske). Muskeldystrofier, Infeksiøs myopati, Toksisk myopati, Metabolsk myopati, Mitokondriemyopati (REV 073, REV 022)
Jan Tore Gran and Øyvind Palm
Kjennetegn på non-inflammatoriske myopatier
Svakhet i muskler uten tegn til inflammasjon.
En gruppe ulike sykdommer av nevrologisk, metabolsk eller annen type som debuterer i ulike aldre og har forskjellige årsaker.
Anamnese og klinisk undersøkelse danner grunnlag for valg av supplerende undersøkelser.
ICD-10: G71.0 Muskeldystrofi; G73.6 (metabolsk myopati); G73.5 (endokrinologisk årsak); G73.4 (Infeksiøs myopati); G72.2 (Toksisk myopati); G13.0 (Malignitet med myopati); G71.3 (Mitokondrium-myopati). G72.0 (Legemiddelutløst myopati)
Kunnskap om non-inflammatoriske myopatier er en viktig del av kompetansen til en revmatolog. Denne kunnskapen er avgjørende for å gi optimal behandling til pasienter med både revmatiske sykdommer og non-inflammatoriske myopatier, for å forebygge komplikasjoner og for å forbedre kommunikasjon og samarbeid med andre spesialister.
Symptomene kan ligne på symptomer på revmatiske sykdommer, noe som kan gjøre diagnosen vanskelig. Revmatologer med kunnskap om non-inflammatoriske myopatier er bedre rustet til å skille mellom disse to tilstandene.
Definisjon
Non-inflammatoriske myopatier er en gruppe muskelsykdommer som ikke er forårsaket av betennelse. De skiller seg dermed fra de inflammatoriske som polymyositt, dermatomyositt, juvenil dermatomyositt (JDM). immunmediert nekrotiserende myopati (IMNM), antisyntetase syndrom og inklusjonslegememyositt. Til forskjell fra disse, forventes ved de non-inflammatoriske ikke antistoff i serum. Non-inflammatoriske myopatier kan skilles ved ulik klinikk, biopsi og ved genetiske undersøkelser. De kan deles inn i hovedgrupper som er beskrevet i avsnittene nedenfor:
- Idiopatiske myopatier (uten kjent årsak)
- Infeksiøs myositt
- Kongenitale myopatier
- Metabolske myopatier
- Mitokondrie myopatier
- Muskeldystrofier
- Sekundær myopati til systemisk sykdom
- Toksisk myopati
Symptomer
Non-inflammatoriske myopatier kan i likhet med de inflammatoriske typene (myositt), føre til muskelsvakhet, gangvansker, svelgevansker og tretthet. De ulike typene kan debutere i ulike aldersgrupper og ha særegne kjennetegn (vennligst se nedenfor).
Undersøkelser
Anamnesen kartlegger eventuell arvelig disposisjon, symptomdebut, symptomer og forløp.
Klinisk gjøres en generell undersøkelse kombinert med vurdering av utseende av muskler (hyper-/hypotrofi) og muskelkraft/fysisk funksjon.
Blod- og urinprøver skal utelukke tegn til infeksjoner, inflammasjon, elektrolyttforstyrrelser, metabolske sykdommer, systemiske bindevevssykdommer og andre tilstander. Blant antistofftester er ANA aktuell for å utelukke tegn til inflammatoriske bindevevssykdommer.
Bildediagnostikk. MR-undersøkelse av angrepet muskulatur kan lokalisere angrepne områder som egner seg for biopsi.
Biopsi fra affisert muskulatur kan i noen tilfeller være helt avgjørende for en sikker diagnose.
Differensialdiagnoser
- Inflammatoriske myopatier i form av polymyositt, dermatomyositt, juvenil dermatomyositt (JDM). immunmediert nekrotiserende myopati (IMNM), antisyntetase syndrom og inklusjonslegememyositt.
- Lysosomale lagringssykdommer (Mukopolysakkaridoser, Mukolipidoser, Oligosakkaridoser, Pompe sykdom, Gaucher sykdom, Fabry sykdom med flere)
- Polymyalgia revmatika
- Fibromyalgi og andre kroniske smertetilstander.
- Nevrogene sykdommer som primært affiserer nerver (sekundært påvirker muskelfunksjonen), for eksempel polynevropati, Parkinsons sykdom, multipel sklerose og amyotrofisk lateralsklerose).
Behandling
Behandlingen av non-inflammatoriske myopatier avhenger av den spesifikke diagnosen og kan variere betydelig. Noen non-inflammatoriske myopatier er genetiske og har ingen kurativ behandling, mens andre kan være ervervet og potensielt reversible.
Målet med behandlingen kan våre å lindre symptomene og forbedre funksjon og livskvaliteten. Behandlingen kan omfatte fysioterapi, ergoterapi og behandling av underliggende sykdommer.
1) Idiopatiske myopatier
ICD-10: G71.9 (uspesifisert)
Definisjon: Idiopatisk non-inflammatorisk muskelsykdom en inhomogen gruppe uten kjent sykdomsårsak. Antall tilfeller blir færre ettersom nyere diagnostikk avdekker stadig flere spesifikke sykdomsmekanismer som klassifiserer dem i de øvrige gruppene ovenfor.
2) Infeksiøse myositter
Definisjon. Infeksiøs myositt skyldes agens som bakterier, sopp, virus eller parasitter.
Symptomer omfatter smerte, ømhet, hevelse og/eller muskulær svakhet. Generelle infeksjonstegn med utmattelse, nattesvette og feber er vanlig, men kan være lite fremtredende ved et svekket immunsystem (for eksempel ved immundempende medikamenter, HIV, høy alder) og ved opportunistiske infeksjoner.
Diagnostisering baseres på kliniske- bakteriologiske og serologiske tester og muskelbiopsi. Selv om molekylær genetisk sekvensering er mulig for infeksiøse agens, inngår dette ikke blant rutine-undersøkelsene (Narajannappa G, 2021).
-Bakterielle myositter
Bakteriell myositt kan defineres som muskelinfeksjon med eller uten dannelse av abscesser. Bakterielle infeksjoner i tverrstripet muskulatur kan skyldes invasjon fra nærliggende infeksjon, penetrerende traume, vaskulær insuffisiens eller hematogen spredning. Immunsupprimerte er spesielt utsatt.

Utløsende agens omfatter S. aureus myositt, gruppe A streptokokk nekrotiserende myositt, gruppe B streptokokk myositt, clostridium infeksjoner (gassgangren), synergistisk non-clostridium myonekrose (spredning fra subcutis og fascier) og aeromonas myonekrose (traume oppstått under kontakt med ferskvann) (Narajannappa G, 2021). Gangren oppstår på grunn av vaskulær insuffisiens/iskemi.
Infeksiøs myositt kan klassifisere i følgende kategorier: –Pyomyositt: Skyldes hematogen spredning og blir ofte omtalt som “tropical myositis”. Hos oss sees dette hos kreftpasienter, immunsupprimerte pasienter, HIV-infiserte, diabetikere og hos pasienter med leversykdommer. Staphylococcus aureus hyppigst, dernest gruppe A streptokokker. Debuterer gjerne over 1-3 uker med lokal hevelse, feber og noe smerte (Nakyama Y, 2021; Crum-Cianflone NF, 2008). –Psoasabscess: Klassifiseres som primær når det ikke kan påvises noen sikker kilde for infeksjon og sekundær når den skyldes spredning fra nærliggende infeksjon. S. aureus vanligst. Pasienter med nedsatt immunsystem som blant eldre, ved diabetes eller ved immunsuppressiv medikasjon er mest utsatt. Klinisk ytrer psoasabscess seg ved feber, halting og smerte (flankene, lyske) og nedsatt allmenntilstand med vekttap (Vajdic ID, 2021; Crum-Cianflone NF, 2008).
-Soppinfeksjoner i muskulatur
Inntreffer vanligvis bare hos immunsupprimerte og sekundært til soppinfeksjon annet sted. Candida, Cryptokokker, Cocci-diodes, Aspergillus og mange andre (Crum-Cianflone NF, 2008).
-Virale myositter
Angriper muskulaturen diffust, uten abscesser. Intense myalgier og uttalt myositt kan ses. Rabdomyolyse er også rapportert. Influenzae A og B, Enterovirus, HIV, type I HTLV (human T-cell leukemia lymphoma virus), hepatitt B og C, parainfluensa, adenovirus, respiratorisk syncytialt virus. Parvovirus (Crum-Cianflone NF, 2008).
-Parasitt betingede myositter
De fleste av disse pasientene smittes med parasitter under opphold utenlands. Trikinose, toksoplasmose, Chagas sykdom (trypanosoma) og andre (Crum-Cianflone NF, 2008).
Litteratur
Chrum-Cianfione. Clin Microbiol Rev 2008
3) Kongenitale muskeldystrofier
ICD-10: G71.2
Definisjon. Kongenitale muskeldystrofier (CMD) er en gruppe arvelige nevromuskulære sykdommer som kjennetegnes ved muskelsvakhet og muskelsvinn som er til stede ved fødselen eller utvikler seg i løpet av de første levemånedene. Disse tilstandene skyldes feil i gener som er involvert i muskelutvikling og funksjon.
Symptomer: Varierende penetrans. Nyfødte med slapp «floppy» muskeltonus. Kan medføre respirasjonsproblemer. Kan være assosiert med malign hypertermi og rabdomyolyse (Pasrija D, 2023). Nomenklatur etter anormale sentrale deler («cores» av muskelfibre (under mikroskop).
Noen eksempler på kongenitale muskeldystrofier:
- Dystroglykanopatier er en gruppe av muskelsykdommer som skyldes defekter i et protein som kalles dystroglykan. Dette proteinet er viktig for muskelcellenes funksjon og struktur. Dystroglykanopatier kan påvirke både muskler og hjerne, og symptomene varierer avhengig av den spesifikke genetiske defekten.
- Fukuyama kongenitale muskeldystrofi (FCMD): Denne formen er en en dystroglykanopati som er mer vanlig i Japan og kjennetegnes av muskelsvakhet, forsinket utvikling og alvorlige hjerneskader.
- Merosin-mangelfull CMD (MDC1A): Denne formen skyldes mangel på proteinet merosin, som er viktig for muskelcellenes struktur. Symptomene varierer, men inkluderer ofte generell muskelsvakhet, forsinket motorisk utvikling og leddkontrakturer. Noen personer med MDC1A kan også ha problemer med hjernen og øynene.
- Muscle-eye-brain sykdom (MEB): Denne formen involverer også både muskler og hjerne. Symptomene inkluderer muskelsvakhet, synsproblemer og kognitive vanskeligheter.
- Nemalin-legeme myopati (NEM) er også en sjelden, arvelig muskelsykdom som kjennetegnes av tilstedeværelsen av nemalin-legemer i muskelfibrene. Disse legemene er unormale proteinkomplekser. Symptomene ligner på CCD, men kan også inkludere vanskeligheter med å puste og spise.
- Sentral kjerne (central core)-sykdom (CCD) er en sjelden, arvelig muskelsykdom som kjennetegnes av en kjernefri sone i midten av muskelfibrene. Symptomene varierer fra mild til alvorlig, men inkluderer ofte muskelsvakhet, forsinket motorisk utvikling og skoliose.
- Ullrichs kongenitale muskeldystrofi (UCMD): Denne formen kjennetegnes av muskelsvakhet, leddkontrakturer (spesielt i knær og hofter) og økt bevegelighet i andre ledd. Noen personer med UCMD kan også ha problemer med hjerte- og/eller lunger.
- Walker-Warburg syndrom (WWS): Dette er en alvorlig form (en dystroglykanopati) for CMD som involverer både muskler og hjerne. Symptomene inkluderer muskelsvakhet, forsinket utvikling, synsproblemer og kognitive vanskeligheter.
Litteratur: Luo s-S L. Muscle Nerve 2010; 43: 402-9; Stensland. Neuromusc Disorders 2011; 21: 41-6; Udd B. Neuromusc Disorders 2012; 22: 5-12; North KN, 2014 (kongenitale myopatier); Bönnemann CK, 2014 (kongenitale myopatier)
4) Metabolske myopatier
ICD-10 G73.6
Definisjon: Metabolske myopatier skyldes oftest en enzymdefekt som rammer muskelenergimetabolismen. Mitokondriesykdom er blant de vanligste metabolske sykdommene og er omtalt i eget avsnitt nedenfor på denne siden. Sykdommene påvises oftest ved biopsi.
Det finnes fire metoder for produksjon av energi (dvs. adenosin trifosfat – ATP) i muskulatur: 1) glykogen metabolismen, 2) lipid stoffskiftet, 3) fosfo-kreatinin lagre og 4) purinnukleotid syklus. I hvile, ved kulde og under faste skjer det hovedsakelig nedbrytning av fettsyrer. Ved submaksimalt arbeid (jogging) skaffes energi først via aerob glykolyse (glykogenolyse / elektrontransport-kjeden), etter hvert fettsyreomsetting. Ved maksimalt arbeid benyttes anaerob glykolyse, fosfokreatinin og ATP via purinnukleotid syklus. Glykogen (brytes ned i cytosolen) brukes altså til muskelenergi ved kraftkrevende arbeid, mens ved lav-aktivitet er lipider hovedkilden for muskelenergi. ATP-produksjon under anaerobe forhold er mindre effektivt og krever nedbrytning av pyruvat til laktat i cytoplasma. All energiproduksjon foregår til slutt i mitokondriene.
Genetikk: Familiær forekomst av muskelsykdom (17%).
Symptomer
Smerte, muskelkramper og muskulær svakhet er de vanligste symptomene. evt. CK-stigning kommer oftest etter anstrengelse og kan være betydelig. Symptomene hos voksne opptrer ofte mange timer etter hard muskelbelastning.
“Second wind”. Spesielt ved defekter i glykogenolysen opptrer smerter etter kortvarig hardt muskelarbeid. Overkommer pasienten imidlertid denne første smerten og kraftsvikten, kan de komme seg igjen gjennom nedbrytning av fett eller mobilisering av glukose fra blodbanen. Dette kalles ofte “second wind”. Dette gjelder de dynamiske metabolske myopatiene, men ikke de statiske metabolske myopatiene som Cori-Forbes, Andersen og Pompe.
Rabdomyolyse kan utvikles etter selv moderat muskelaktivitet.
Andre symptomer. Myopati kan være eneste symptom eller kombinert med symptomer som retinopati, cerebrale, polynevropati og leveraffeksjon i noen av tilfellene.
Undersøkelser
Anamnesen kartlegger eventuelle familiære tilfeller (genetikk), symptomdebut og symptomer, samt vurdering av utløsende faktorer.
Klinisk gjøres en generell indremedisinsk status og nærmere undersøkelse av muskulatur (hypo-/hypertrofi, kraft-tester)
Laboratorieprøver: Hb, leukocytter med differensialtelling, trombocytter, blodsukker, Na, K, Ca, ASAT, ALAT, CK (CK-forhøyelse hos 57 %), laktat, frie fettsyrer, 3-hydroksybutyrat, total og fri karnitin, myoglobin, TSH, f-T4. Differensialdiagnostisk ANA og myosittspesifikke antistoff, ACE.
Ofte er total karnitin lav og ratioen frie fettsyrer/ 3-hydroksybutyrat høy (nedsatt laktogenese). Ved defekter i fettsyrenedbrytningen vil de biokjemiske avvik ofte være påvisbare bare ved symptomer eller ved belastning.
Ved forstyrrelser i glykogenolysen sees CK-stigning ved sykkelbelastning.
«Forearm ischemic test» gir manglende laktatstigning ved alle glykogenolyser bortsett fra ved fosforylase kinase b-mangel, Glykogen brancher enzymsykdom (Andersen’s disease, amylopectinosis) og maltase-mangel.
| Sykdom | Laktat | Ammoniakk |
| Normalt | Stiger 3-4 x normalen | Stiger 3-4 x normalen |
| Inkonklusiv test | Ingen stigning | Ingen stigning |
| Type III, V, VII, IX, XI | Ingen stigning | Stiger 3-4 x normalen |
| MAD (Multippel acyl-CoA dehydrogenasemangel) | Stiger | Ingen stigning |
β-oksidering av frie fettsyrer skjer anaerobt i mitokondriene og genererer ATP. Korte og mellomlange fettsyrer passerer fritt over fra cytosolen til mitokondriene. Lange fettsyrer derimot må først bindes til karnitin, en prosess som involverer acyl-karnitin-translokase og karnitin palmitoyltransferase (CPT) I (over ytre membran av mitokondriene) og II (over indre membran).
Muskelbiopsi. Økte glykogen- eller lipidavleiringer ved histologisk u.s
Sykdommer relatert til forstyrrelser i glykolysen (Glykogenavleiringssykdommer) (Stone WL, 2021)
ICD-10 G73.6
Det er 4 typer metabolsk myopati som nesten alltid debuterer med muskelsymptomer: type V, VII, X og Xl. De andre kan også starte med manifestasjoner fra andre organer.
Type 0. “Muskel-aglykogenose”.
Her skjer det ingen avleiring av glykogen fordi det ikke syntetiseres. Skyldes mangel på enzymet glykogen syntetase. Defekt glykogen syntese kan imidlertid også skyldes mangel på muskel-glykogenin som er det enzymet som setter sammen de første glykosyl-enhetene i starten på syntesen av glykogen.
Type IA. von Gierkes sykdom (glukose-6-fosfatase-mangel).
Gir egentlig ingen myopati, men glykogenavleiring i lever.
Type II. Alfa-glykosidase-mangel (Pompe sykdom er den infantile form).
- Voksen type har debut etter 20 års alder. Ofte proksimal, men 1/3 debuterer med respirasjonssvikt. Hypertrofi av legger. Muskelbiopsi viser atrofi og glykogen-overskudd.
- Vennligst se også kapitlet av lysosomale lagringssykdommer
Type III. Cori-Forbes sykdom (amylo-1,6 glykosidase-mangel).
Debuterer med leverdysfunksjon i barnealder. Remisjon omkring puberteten. Myopati med muskelsvakhet i 30-40 års alder. Atrofi av distal muskulatur i underekstremitetene og intrinsike håndmuskler. Som oftest relativt mild sykdom. Type III a rammer lever og muskel, type III b bare lever.
Type IV.“Branching” glukosidase-mangel (Andersens sykdom).
Bare små mengder glykogen avleires, men disse har lange sidegreiner. Rammer barn. Fatal leversvikt. Ev. hjerte og hjerne.
Type V. McArdles sykdom (myofosforylase-mangel). (Vieitez I Neuromusc Disord 2011).
Genetikk: Autosomal recessiv arv (mutasjoner i PYGM-genet som koder for myofosforylase).
Prevalens: 1/100 000. Mann : kvinne ratio er 1. Debuterer i barnealder (alvorlig) eller i voksen alder.
Patogenese: Myofosforylase starter nedbrytningen av muskelglykogen ved å omdanne glykogen til glukose-1-fosfatase. Imidlertid forstyrres ikke muskelfibrenes evne til å oppta glykose fra blodet slik at energiproduksjonen ikke blir fullstendig hemmet.
Epidemiologi: 50 % er mellom 10-20 år.
Symptomer: Fysisk belastning etterfølges av myalgi, tretthet, stivhet og svakhet i anvendt muskulatur. Dette skjer gjerne ved intense isometriske belastninger (vektløftning bl.a.). Ofte krampeliknende plager som først reduseres etter timer. Skyldes muskelnekrose og myoglobinuri. Barn får ofte fatigue, tenåringer ofte kramper og muskelsvakhet, mens hos voksne dominerer proksimal muskelsvakhet. Risiko for utvikling av akutt nyresvikt.
Laboratorieprøver: CK oftest forhøyet, også utenfor anfall.
Kraft-test: Ganske typisk for McArdles sykdom er “second wind” fenomenet. Pasientene erfarer plutselig en reduksjon av plagene (muskelsvakhet, fatigue, tachykardi) etter omkring 10 minutters belastning. Dette skyldes at i løpet av de første minuttene av moderat belastning skjer det en vasodilatasjon i muskelfibrene som frigjør mer glykose til disse.
Ved muskelsmerter som opptrer under fysisk belastning, skal pasienten rådes til å stoppe opp (ellers fare for rabdomyolyse). Forsiktig med statiner til disse pasientene (kan utløse rabdomyolyse).
Behandling: Lav dose kreatin (60 :mg/kg i 4 uker) kan forsøkes. Dietten skal være rik på komplekse karbohydrater (frukt, pasta, kornprodukter). Man kan også forsake inntak av 30-40 g glykose 5 minutter for start av fysisk krevende arbeid.
Type VI. Hers sykdom (lever-fosforylase-mangel). Leveraffeksjon dominerer.
Type VII. Taruis sykdom (muskel-fosfofruktokinase-mangel). Myopati og hemolytisk anemi, evt. arthritis urica. Anstrengelsesintoleranse. CK kan være forhøyet.
Type VIII. Phosphorylase B kinase-mangel. Barn. Leverforstyrrelse og/eller myopati. Voksne har progredierende muskelsvakhet.
Type IX. Fosfoglycerat kinase mangel. X-bunden. Debuterer med hemolytisk anemi og CNS-affeksjon (kramper og mental retardasjon). Kan gi myopati.
Type X. Fosfoglycerat mutase-mangel. Anstrengelses-intoleranse, muskelkramper og tilbakevendende myoglobinuri.
Type XI.Laktatdehydrogenase mangel (Fanconi-Bickel syndrom). Debut første lever.
Type XII.Aldolase A-mangel. Barn.
Type XIII.Triosefosfatase isomerase mangel.
Type XIV.Fosfoglukomutase mangel. Småbarn, muskelkramper ved aktivitet.
Type XV. β-enolase mange, (myalgi). Anstrengelses-intoleranse, tilbakevendende myoglobinuri. Eksem hos noen.
Sykdommer relatert til forstyrrelser i lipidmetabolismen (“Lipid storage myopathy”) (Liang W-C, 2010)
ICD-10 G73.6
Medfødte defekter i fettsyrenedbrytningen vil ofte manifestere seg som multiorgansykdom når de utvikles i barnealderen, mens ved start i voksen alder kan myopati være eneste kliniske funn.
Patogenese: Defekte enzymer i B-oksyderingen av frie fettsyrer gir forstyrrelser i transporten av fett over mitokondriemembranen. Karnitin spiller en viktig rolle for denne transporten. Økte fettavleiringer i muskulatur sees ofte, men kan også forekomme ved inaktivitet og andre muskelsykdommer.
Symptomer: En tommelfingerregel er at smerter- og ev. muskelsvakhet eller tretthet ofte debuterer mange timer etter hard muskelbelastning, faste eller utsettelse for lave temperaturer.
Laboratorieprøver: Dersom symptomene opptrer anfallsvis, bør blodanalysene tas under en akutt episode og omfatte: glukose, ALAT, CK, laktat, frie fettsyrer og 3-hydroksybutyrat, total og fri karnitin og myoglobin. Urin bør sendes til “metabolsk screening”.
Dersom analysene er normale, men den kliniske mistanken fortsatt til stede, gjentas analysene etter 24 timers faste. Ved disse typene av metabolsk myopati vil total karnitin ofte være lav med redusert fri fraksjon og ratioen frie fettsyrer/3-hydroksybutyrat høy (>3).
Karnitin-mangel: Både primære og sekundære former (Fanconis syndrom, hemodialyse og valproat behandling). Hovedsakelig tre fenotyper: kardiomyopati, myopati og hypoketotisk hyperglykemi.
Sykdommer med massiv lipidose
Nøytralt lipid avleiringssykdom (NLSD) rammer trigyserider. Svakheten utvikles i annen og tredje dekade, proksimal og distal muskulatur
Multippel acyl-CoA dehydrogenasemangel (MAD)
Sykdommer med beskjeden lipidose
“very-long-chain” acyl-CoA dehydrogenase-mangel (VLCAD)
“Mitochondrial trifunctional” protein-mangel (MTP)
“Phosphatide acid phosphatase” mangel (LIPIN)
“Medium chain acid” CoA dehydrogenase-mangel (MCAD)
“Short chain acid” CoA dehydrogenase-mangel (SCAD)
Litteratur: Laforet P &Vianey-Saban C. Neuromusc Disord 2010
Karnitin paimitoyltransferase-mangel (CPT II) (Lehmann D, 2017)
Disse enzymene assisterer i transporten av lange fettsyrer over mitokondriemembranen slik at de senere kan oksideres.
Tre typer: Neonatal, infantil og sent debuterende type. Den siste affiserer kun muskulatur. Ikke uvanlige tilstander, debuterer gjerne hos yngre voksne med muskelsmerter og mørk urin etter fysiske belastninger (myoglobinuri sees hos 80 %). Kan utvikle rabdomyolyse. Utløsende faktorer utover langvarig fysisk belastning er infeksjoner, faste, diazepam og ibuprofen. CK ofte normal utenfor anfall. Behandling: I-karnitin og unngå faste.
- Vennligst les om metabolske tilstander og revmatologi i eget kapittel
5) Mitokondrie-myopatier/mitokondriesykdom
ICD-10 G71.3
Historie: Den første mitokondriesykdom (Lufts sykdom) ble rapportert i 1959, fire år før oppdagelsen av mitokondrie DNA.
Patogenes og genetikk: All energiomsetning i cellen foregår i mitokondriene (Krebs syklus, respirasjonskjeden, fettoksydasjon, pyruvat oksydasjon, fettransport). Mitokondrielt DNA (mtDNA) arves fra mor (maternell arvegang), slik at familie-anamnesen er viktig ved utredning av denne type sykdom. mtDNA har høy frekvens av mutasjoner som resulterer i at cellen ofte har både normalt mtDNA og mutert mtDNA. Dette kalles heteroplasmi. Det kreves ofte 60-90% mutert mtDNA for kliniske symptomer utvikles. Polymerase gamma (PLOG) er et enzym som replikerer og reparerer mitokondrie-DNA. Mutasjoner i genet som koder for den katalytiske delen av enzymet, PLOG-genet, er en av de hyppigste årsakene til mitokondriesykdom. Mitokondrie-sykdommer kan også skyldes mutasjon i kjerne-DNA og arves da etter mendelsk arvegang.
Forekomst: Antallet kjente mitokondrie sykdommer har økt de siste årene og regnes, samlet sett, som den hyppigste medfødte metabolske sykdommen. Blant de vanligste er Leigh syndrom og MELAS (vennligst se nedenfor). Det er beregnet at 1:10.000 barn i Sverige utvikler mitokondrie-sykdom før skolealder (Darin N, 2001), men symptomene kan debutere i alle aldre.
Symptomer: Mitokondrie-sykdom kan ramme flere organsystemer. Sykdomsbildene og alvorlighetsgrad varierer mellom de ulike formene og fra person til person og aldersgrupper. Revmatologisk fremstår tap av muskelstyrke og hypotone muskler som ses i alle aldre. Andre mulige manifestasjoner er forstyrret syn-, hørsel-, hjerte-, lunge-, lever, nyre- og påvirket tarmfunksjon (pseudoobstruksjon). Noen får læringsvansker og alvorlige nevrologiske og/eller psykiske symptomer. Hos små barn kan symptomene være vanskelige å tolke (forsinket utvikling, redusert vekst, hypotone muskler, avtakende fysisk funksjon).
Isolert muskelsykdom forekommer. Typisk er da redusert toleranse for anstrengelse og trening. Utmattelse, muskelsvakhet, muskelsmerter og påfølgende stigning i enzymet CK (kreatin kinase) i blodprøver.
Mitokondrie myopati forårsaket av mutasjon i mtDNA:
- Familiær tilbakevendende myoglobinuri
- Kearn-Sayre syndrom (kronisk progressiv ekstern oftalmoplegi)
- Lebers hereditære optiske nevropati (LHON)
- Leigh syndrom (Subakutt nekrotiserende encefalomyopati)
- MELAS (mitokondriell encefalopati, melkesyreacidose og slag-liknende episoder) (Varhaug KN, 2022)
- MERFF (myoklonisk epilepsi med ragged red fibers) m.m.
- Mitokondriell myopati forårsaket av nukleære mutasjoner: MNGIE (mitokondriell nevrogastrointestinal encefalomyelopati)
- NARP (nevrogen svakhet, ataksi, retinitis pigmentosa)
- Pearsons sykdom
- PEO (progressiv ekstern oftalmoplegi)
Blodprøver: Forhøyet laktat forekommer i blod og ca. 70% har forhøyet protein i spinalvæsken. Blodprøver skal omfatte celle-tellinger, lever- og nyrefunksjonsprøver. Biomarkører for mitokondriesykdom som rammer muskulaturen er fibroblast-vekstfaktor 21 (FGF-21) og vekstdifferensieringsfaktor 15 (GDF-15).
MR caput og EEG gjøres ved epilepsi eller andre CNS-symptomer.
Biopsi: Ved muskelbiopsi ses “ragged red fibres” (noen få kan normalt sees hos eldre, differensialdiagnose: Inklusjonslegememyositt), og de irregulære monocyttene farges rødaktig ved Gomorri-farging.
Diagnosen: Spesifikk diagnostisering gjøres ved funn av mutasjoner. En screener først for de fire vanligste (founder-mutasjoner). Full genetisk sekvensering kan gjøres ved negativt funn, men likevel sterk mistanke om mitokondrie-sykdom.
Differensialdiagnoser: Myositt, muskeldystrofi, amyotrofisk lateralsklerose (ALS), multippel sklerose (MS) og andre nevrogene, progressive sykdommer, antifosfolipid syndrom og andre disposisjoner for slag.
Behandling: Tverrfaglig tilnærming basert på hvilke symptomer den enkelte har. Symptomlindring (muskelsmerter, kramper, diabetes, hjertesvikt, hørselstap) er viktig. Tilpasset fysisk aerob trenging/øvelser. Kombinasjon av høye doser koenzym Q10, i kombinasjon med kreatin og L-karnitin kan forsøkes, men dokumentasjon av effekt på slike vitamin/kostholdstilskudd er ikke entydig.
Litteratur:
6) Muskeldystrofier (arvelige myopatier)
ICD-10 G71.0
Definisjon
Muskeldystrofier er en heterogen genetisk betinget sykdomsgruppe som klinisk karakteriseres av progredierende proksimal muskelsvakhet. Sykdommene skyldes utilstrekkelig eller manglende glykoproteiner i muskelcellenes plasmamembran (Allan DG, 2015). Ved muskelbiopsi påvises dystrofi, hvilket innebærer degenerasjon og regenerasjon av muskelfibre. I tillegg sees fibervariasjon og sentralt lokaliserte myonuclei. Med tiden erstattes muskelvevet av fett og fibrøst vev. CK er ofte forhøyet. En kjenner til over 30 typer muskeldystrofi. Hver type muskeldystrofi har sitt genetiske arvemønster og defekter i de spesifikke gener medfører de ulike sykdomsmanifestasjonene (La Pelusa A, 2023). Mange debut i barnealderen, men start i voksen alder forekommer og da gjerne med myopati som dominerende symptom. Debutalder kan gi viktige diagnostiske holdepunkter (Guzman OdRC, 2012). I løpet av de senere årene er mange av de tilhørende gen-defekter kartlagt (genportalen.no).
Sykdomsårsak
Sykdommene skyldes DNA mutasjoner hvor delesjoner gir tap av DNA, mens punktmutasjoner gir forandringer i koden. Mutasjonene affiserer proteiner lokalisert til muskelcellen. For å forstå de ulike muskeldystrofiene er kunnskap om disse helt nødvendig.
- Dystrofin-glykoprotein kompleks: Dystrofin: lokalisert til den cytoplasmatiske siden av cellemembranen (muskelceller og hjertemuskelceller). Den amino-terminale delen fester seg til aktin (kontraktile element), mens en del fester seg til 13-dystroglykan (transmembran). alfa-dystroglykan er lokalisert ekstracellulært og bindes til alfa-laminin (merosin). Syntropin kompleks (cytoplasmatisk). Festes til dystrobrevin og dystrofin. Sarkoglykan kompleks (cytoplasmatisk) lokaliseres sammen med sarkospan.
- Merosin: Samlenavn for ulike lamininer. Bindes blant annet til alfa-dystroglykan (struktur stabilitet). Forekommer også i perifere nerveceller
- Integriner: Transmembrane reseptorer som sikrer binding mellom ECM og celleskjelettet. Bindes også til merosin.
- Proteiner i cellekjernen: Emerin lokaliseres på innsiden av membranen på skjelettmuskelceller, hjerte- og glatte muskelceller. Laminin A, B og C. Valosin inneholdende protein (VCP) og PABN1
- Enzymer: Kalpain-3 (proteolytisk). Fukutin (glykosyltransferase). TRIM32, GNE
- Andre strukturer: Dysferlin (skjelettmuskel og hjerte). Caveolae er invaginasjoner i sarkolemma (cellemembranen)
Symptomer
Muskeldystrofiene har ulike kliniske symptomer og undersøkelsesfunn som omfatter pseudohypertrofi (store muskler, men redusert muskelstyrke), reduserte respirasjonsmuskler, svak rygg, arytmier, distal muskelatrofi, redusert ansikts- og skuldermuskulatur. Progredierende symptomer, noen fra barne-alder, men flere typer begynner i 20-40 årene (Guzman OdRC, 2012).
Ulike muskeldystrofier:
Limb Girdle Muskeldystrofi (LGMD) (Roche CT, 2010)

Sykdomsårsak. Autosomal recessive former dominerer, men det er funnet autosomalt dominante typer (sjeldne). Omtrent 90 % utgjøres av type 2 (se listen nedenfor). Type 1A og 2B kan ha inflammatoriske infiltrater, mens type 1A, 2G og 2J kan ha “rimmed vacuoles” (differensialdiagnose: Inklusjonslegeme myositt).
Debutalder er variabel.
Undersøkelsesfunn. Både ekstremitetsmuskler og truncus kan angripes.
- LGMD1A: Skyldes mutasjon av genet som koder for myotilin. Ekstrem sjelden. Kan starte i voksen alder. Progressiv muskelsvakhet. Kontrakturer og kardiomyopati kan sees. Dysartri. Histologisk kan “rimmed vacuoles” og inklusjoner påvises.
- LGMD1B: Skyldes mutasjoner i genet som koder for lamin (LMNA). Laminer er filamenter lokalisert til den indre delen av cellekjernen (Se skjematisk tegning). Mutasjoner i LMNAgenet er, utenom limb girdle muskeldystrofi, også assosiert med flere sykdommer, deriblant Emery-Dreifuss muskeldystrofi, Charcot-Marie-Tooth sykdom, familiær lipodystrofi og dilatert kardiomyopati.
- LGMD1C: Skyldes mutasjoner i genet som koder for Caveolin-3. Dette er et protein lokalisert til de såkalte “små huler”, dvs. små invaginasjoner på plasmamembranen hvis funksjon er å delta i cellens signalsystem. Begynner i barnealder eller i voksenalder. Progressiv muskelsvakhet eller myalgi utløst av fysisk anstrengelse. Hypertrofi av legger. Muskelbiopsi viser redusert farging for caveoiin-3.
- LGMD1 D-F: Mutasjoner: DnaJ homolog subfamily B member 6, Desmin, Transportin-3.
- LGMD2A: Disse kalles ofte kalpainopatier. Kalpainer er kalsiumavhengige cystein proteaser. Sykdommene debuterer oftest mellom 8 og 15 års alder med progressiv atrofi av posteriore muskler i ekstremitetene (adduktorer, semimembranosus og vastus intermedius). Abdominalmuskler kan affiseres. Scapulær “winging” hos 80%. Kontrakturer utvikles hos 67%. CK økt opptil 20 ganger normalen. Utgjør 10-30 % av alle LGMD2. Histologisk sees lobulære fibre. Mutasjon av calpain-3. Kan gi asymptomatisk forhøyelse av CK, dog er de fleste ikke gangbare i 40-arsalderen.
- LGMD2B: Disse kalles dysferlinopatier. Dysferlin er et sarkolem protein. Tilstanden rammer oftest unge voksne i annen og tredje dekade (11-48 år). Distal affeksjon kan forutgå proksimalt engasjement med mange år. Biopsi viser ofte inflammasjon. Utgjør 30 % av alle LGMD2. Starter i leggene (“Miyoshi myopati”). Pasienten har vansker med å stå på tærne. CK tydelig forhøyet. Kan diagnostiseres ved å påvise mangel av dysferlin i leukocytter (Western blot).
- LGMD2C-F: Benevnes sarkoglykanopatier. Utgjør 6-10% av alle LGMD2. En type rammer Dystrofin som er en integrert del av muskelcellens cytoskjelett. Hele komplekset kalles DGC (Dystrofin glykoprotein kompleks) og representerer en fysisk forbindelse mellom aktin (kontraktilt element), cytoskjelettet og ekstracelluIær matriks (ECM). Sannsynlig hovedfunksjon er å beskytte sarkolemma mot skader under kontraksjon og relaksasjon. 2D kalles dysferlinopati. Høy CK, evt. også makroglossi.
- LGMD2G: Mutasjoner i genet som koder for telethonin. Rapportert fra Brasil.
- LGMD2H: Mutasjoner i genet som koder for Trim32. Rapportert fra Canada.
- LGMD2I: Mutasjon i genet som koder for FKRP (Fukutin relatert protein). Ikke sjelden i Nord-Europa. Omtrent 50 % utvikler kardiomyopati og svakhet av respirasjonsmusklene. FKRP er lokalisert til endoplasmatisk retikulum (Fukutin til Golgi-apparatet).
- LGMD2J: Mutasjoner i genet som koder for titin. Finland.
- LGMD2: Mutasjon i POMTI-genet (som dog oftest gir Walker-Warburg syndrom). Tyrkia og England.
- LGMD2L: Mutasjon i genet som koder for Fukutin. (Prevalens i Norge 1/54 000).
- LGMD2M: Mutasjoner i POMGnT1 genet (gir oftest Muscle-Eye-Brain sykdom).
- LGMD2N: Mutasjoner i POMTZ.
Dystrofinopatier
-Duchennes muskeldystrofi (Venugopal V, 2023)
Sykdomsårsak: X-bundet recessiv. Defekt i dystrofingenet (X-kromosom), delesjon, spontane mutasjoner hos 1/3.
Forekomst: Debut blant gutter i 2-4 års alderen. Prevalens 1/18 000 menn, debuterer oftest i barneårene,
Symptomer: Redusert kraft i nakke-fleksorer, vaggende gange, tendens til å gå på tå, falltendens,
Undersøkelsesfunn: Hypertrofi av legger, proksimal kraftsvikt, positivt Gowers tegn.
Blodprøver: CK 50-100 ganger forhøyet ved fødselen. Immunohistokjemisk påvisning av manglende eller redusert dystrofin.
Behandling: Ingen effekt av immunsuppressiva
Prognose: Mange sitter i rullestol for 12 års alderen.
-Beckers muskulær dystrofi (Thada PK, 2024)
Genetikk: X-bunden recessiv (gutter affiseres). 10 % skyldes spontane mutasjoner. Defekt i dystrofin-genet (“mild form for Duchenne”).
Forekomst: Insidens 5/100 000 for 20 års alderen.
Symptomer: De fleste får vanskeligheter med forflytning. 28 % debuterer med “legg-klaudikasjon”. Myalgi og muskelkramper. Kardiomyopati forekommer. Ofte redusert IQ.
Blodprøver: CK forhøyet 20-200 ganger øvre normalområde.
-X-bunden dilatert kardiomyopati (Kamdar F, 2016)
Andre:

Fascio-scaplulo-humeral dystrofi (FSHD) (Fecek C, 2021)
Symptomer: Asymmetrisk, progredierende muskelatrofi i ansikt (nedsatt mimikk), skulderregion (vansker med å løfte armer). Kan også angripe muskler omkring hofter. Vinge-scapula (Scapula alata). Senere distal atrofi i ekstremiteter. Hørselstap. Retinale teleangiektasier. Stor variasjon i debutalder og symptomer gjør at tilstanden lange kan overses.
Forekomst (prevalens): Omtrent 4/100.000
Diagnostikk gjøres ved gentest (FSHD type 1 utgjør 95% og er monogenetisk, FSHD type 2 er digenetisk med forandringer i to gener). CK i blod kan være normal eller lett forhøyet. MR viser ofte typisk fordeling av muskel-manifestasjoner med ødem og atrofi. Biopsi skiller fra myositt.
Behandling. Fysioterapi og smerte-kontroll er sentrale i behandlingen.
Skulder-hofte muskeldystrofi
Dystrofia myotonica, DM 1 og DM 2 (Vydro DG, 2023)
Forekomst: 1,5-5,5/100.000
Symptomer: Distal muskelsvakhet i DM1. Proksimalt i DM2. Klinisk myotoni med vansker å relakser etter kontraksjoner. Karatakt, diabetes, håravfall frontalt, arytmier, cholecystitt. Ptose. Svangerskaps-komplikasjoner.
Periodiske pareser (Statland JM, 2017)
Periodisk paralyse skyldes arvelig myopati. Symptomenes alvorlighetsgrad varierer, slik at ikke alle med genetisk disposisjon blir syke. .Autosomal dominant arvegang
- Hypokalemisk periodisk paralyse
- Hyperkalemisk periodisk paralyse
- Kongenital Paramyotoni (ofte sammen med hyperkalemisk periodisk paralyse)
- Andersen-Tawil syndrom (langt QT syndrom
Bent spine/Dropped head syndrom (Peng Y, 2015)
Kan starte etter 60 års alder. Dropped head syndrom forårsakes av svakhet i nakke-ekstensor og har mange mulige årsaker: Motor nevron sykdom (ALS), myastenia gravis, kronisk demyeliniserende polynevropati og andre nevropatier, samt inflammatoriske og metabolske myopatier. I enkelte tilfeller koeksisterer «Bent spine» på grunn av abnormal fleksjon i truncus-muskulatur. Acyl-CoA dehydrogenase mangel (MADD) er en av flere årsaker
Okulo-faryngeal muskeldystrofi
Forekomst: Relativt sjelden. Starter i 40-50 årene.
Symptomer: Ptose og dysfagi. Oftalmoparese og bulbær svakhet med dysartri og dysfagi. Sjelden distal muskelsvakhet.
Ullrich syndrom: (mutasjoner i genet for kollagen VI) (Stavuris J, 2020)
Emery-Dreifuss muskeldystrofi (Heller SA, 2020)
Genetikk: Emerin genet. X-bundet, recessiv
Symptomer: Debut blant gutter I tenårene. Muskelsvakhet, særlig skuldre og legger. Arytmi. Gradvis utvikling av kontrakturer.
Diagnose: Forhøyet CK i blodet. Gentest
Amyloid myopati av familiær årsak
Genetikk: Transthyretin mutasjon. Autosomal dominant arvegang
Utredning: Elektroforese med immunfiksering og kvantitering av monoklonale proteiner i serum og urin
Artrogryposis multiplec congenita er en gruppe medfødt genetiske tilstander som kjennetegnes ved ledd-kontraktur i to eller flere kroppsregioner (Dahan-Oliel N, 2019). En presis diagnose kan gjøres ved genetisk screening i en del av tilfellene (Rustad CF, 2022).
Distale myopatier (noen av disse klassifiseres av noen som IBM) (Dimackie MM, 2014)
Welanders distale myopati
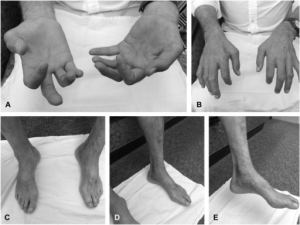
Symptomer: Autosomal dominant, debutalder 20-77 år, noe flere menn, starter oftest i hender (finger-fleksorer og håndledd), svakhet og atrofi, kuldefølelse i hender og føtter hos 90%.
Genetikk: Assosiert med defekt i TIA1 genet som er med å styre apoptose. Andre mutasjoner i TIA1 genet forårsaker amyloid lateral sklerose (ALS) og fronto-temporal demens.
Tibial muskel dystrofi (distal myopati med anterior onset, UDD myopati, Markesbery-Griggs distal myopati)
Genetikk: Autosomal dominant
Symptomer: Debut etter 35-årsalder, initialt m. Tibialis anterior. Aldri håndmuskulatur.
Sen adult form: Autosomal dominant, begynner i underekstremitetene distalt – for så å affisere hender.
Tidlig adult form: Debut 19-20 år. Starter distalt – ofte i m. gastrocnemius, ofte negativ familie-anamnese, CK nesten alltid forhøyet.
Undersøkelser: CK lett forhøyet hos 62 %. Elektron mikroskopi viser ofte vakuoler.
Nonakas distale myopati GNE-myopati. Inklusjonslegeme myositt 2.
Symptomer: Debut fra tenårene til 20-30 årene, fremre partier av leggen. Drop-fot på grunn av muskulær svakhet.
Genetikk: Autosomal recessiv eller autosomal dominant former
Prognose: Oftest rullestolavhengighet fra 20-års alder.
Myofibrillære myopatier
En gruppe morfologisk lignende myopatier som har ulike genetiske årsaker.
Symptomer: Langsom progredierende muskelsvakhet i proksimal og / eller distal muskulatur. Distal affeksjon hos 80%. Enkelte får sensorisk nevropati (20%), muskelstivhet, smerter og kramper. Kardiomyopati hos 15-30%
Miyoshi distal muskelatrofi

Dysferlin-defekt som også er assosiert med limb girdle dystrofi type 2B (LGMD2B) som imidlertid preges av proksimal svakhet. Miyoshi dystrofi debuterer oftest omkring 19 års alder. Muskulær atrofi og svakhet, særling i legger (m. gastronemicus og m. soleus). Over noen år angripes også lår- og gluteal muskulatur. Delvis også underarmer, men aldri hender.
Nemalin myopati (NM) 3 (Jirka C, 2019)
Svakhet, hypotoni og utslukkede dype senereflekser. Ansikt, nakke-fleksorer, proksimal ekstremitetsmuskulatur. I alt seks typer, men overlapp forekommer.
- Alvorlig kongenital (neonatal) (16%)
- Intermediær kongenital (20%)
- Typisk kongenital form (46%)
- Barnealder-debut(13%)
- Voksen-debut (late-onset) (4%)
7) Myopati sekundært til annen sykdom
ICD-10; G73.7

Muskelfunksjonen lider på grunn av annen sykdom, mangelfull ernæring eller alkoholisme.
Endokrin myopati
ICD-10 G73.5
Thyreoidea-sykdom, Parathyroidea-sykdom, hypofyse eller binyre-sykdom
Thyreotoksikose: Muskelsvakheten lokaliseres oftest proksimalt og er mer uttalt enn hva atrofien skulle tilsi. Bulbære muskler og øsofagus kan affiseres (akutt thyreoid myopati). CK oftest normal. Tilstanden bedres ved neomercazol. Thyreotoksisk periodisk paralyse er tilbakevendende episoder av muskelsvakhet som varer fra minutter til dager. Kan utløses ved avkjøling eller fysisk belastning. Ofte lav CK under anfall (Lin S-H, 2012).
Hypotyreose: Symptomer er proksimal svakhet, tretthet, langsomme bevegelser av musklene og myalgi. Laboratorieprøver kan vise høy CK. Kan debutere med rabdomyolyse. Mildere former er vanlig (Farrduddin MM, 2023).
-Hoffman syndrom (adult hypotyreoid myopati med smertefulle kramper).
-Kocher-Debre-Semelaigne syndrom (Hoffman syndrom uten kramper). Nedsatte reflekser.
Akromegali (høy somatotropin/veksthormon): 50 % får muskelsvakhet. Gradvis og progressiv. Lite atrofi. CK lett økt.
Hyperparatyroidisme: Proksimal muskelsvakhet. CK ofte normal. EMG viser nedsatt størrelse av motor-unit potensialene og økte polyfasiske potensialer uten spontan aktivitet (Bilezikian JP, 2016).
Hypoparatyreose: Distal nummenhet, parestesier, carpopedal spasme, diffuse muskelkramper (Chvosteks og Trousseaus tegn ofte positive).
Steroid myopati (og Mb. Cushing): Vennligst se Toksisk myopati ovenfor
Mb. Addison: Mellom 25% og 50 % av pasienter med binyresvikt har muskelkramper og svakhet: dette gjelder uansett årsak til binyrebark-svikten. Behandles med kortison. Kan også gi hyperkalemisk periodisk paralyse.
Hypopituitarisme / hypofyse-svikt (Simmond-Sheehans syndrom): Uttalt svakhet med atrofi.
Systemisk inflammatorisk sykdom
Mixed connective disease (MCTD): Myositt inngår i klassifikasjonskriterier og ses hos ca. 30%.
Revmatoid artritt: Myopati er sjelden, utenom medikament-betinget (toksisk).
Sjøgrens syndrom: Myositt hos 3%
Systemisk lupus erythematosus (SLE): Myopati oftest relatert til behandling med steroider eller hydroksyklorokin (Plaquenil).
Systemisk sklerose: Skleromyositt med ant-Scl 75/100 antistoff (vennligst se systemisk sklerose).
Elektrolytt-forstyrrelser
Kalium eller magnesium (hypo- og hyper-). Hyperkalemisk periodisk paralyse (Sekkon DS, 2023).
- Hypokalemi: lett: 3,0-3,5mml/L (uspesifikke eller ingen symptomer), moderat 2,5-2,9 (slitenhet) , alvorlig <2,5mm/L (risiko for hjertearytmi og pareser)
Hypofosfatemi
Critical illness myopati (Sheperd S, 2017)
Non-depolariserende nevromuskulær blokkerende stoffer
Steroid-myopati
Amyloid myopati
8) Toksisk / medikament-utløst myopati
ICD-10: G72.2 (Toksisk myopati)
Blant toksiske myopatier utgjør medikament-utløst myopati den største gruppen. Symptomene kommer oftest uker eller måneder etter oppstart og bedres få uker etter seponering av utløsende agens. Vær spesielt oppmerksom på bruk av statiner, sjekkpunkthemmere, kortikosteroider, hydroksyklorokin (Plaquenil) og kolkisin.
Medikamenter som kan utløse muskelsmerter (myalgi): Nalidiksinsyre (fluorokinoloner mot urinveisinfeksjon), ciclosporin, klofibrat, statiner, levodopa, prokainamid, hydralazin, fenytoin, barbiturater, enalapril, karbimazol, metoprolol.
Myopati (myalgi og myositt) ved lipidsenkende medikamenter (statiner): Vennligst les om Immunmediert nekrotiserende myopati (IMNM) i kapitlet om myositt
Kolkisin-myopati: Ofte også perifer myopati. Vakuole-myopati i type I fibre (differensial-diagnose: inklusjonslegeme myositt).
Klorokin og hydroksyklorokin-myopati: Begynner nesten alltid i underekstremitetene. “Curvilinnear bodies”
Myotoni-lignende: Propranolol
Myasteniforme: Penicillamin, klorokin, betablokkere, tetrasykliner, litium, morfin.
Tendinopati/seneruptur: Dette kan sees etter bruk av antibiotika (fluoroquinolon) og skyldes induksjon av metalloproteinaser. Også ved kortikosteroider (systemisk og lokalt)
Hypovitaminose D: kan medføre mange symptomer, deriblant muskelsvakhet
Lokal myotoksisitet: Ved gjentatte i.m. injeksjoner (oftest når samme injeksjonssted benyttes) av anestetika kan lokal nekrose oppstå (Nicolaus syndrom), evt. kontrakturer.
Malign hypertermi: Utvikles typisk hos genetisk disponerte individer når de utsettes for generell anestesi. Høyfebrilia, metabolsk acidose, muskelrigiditet, Stigende CK, myoglobinuri (urin myoglobin > 12pg/m1). Disponerende: Trisykliske anti-depressiva, monoaminooxidase-hemmere (Watt S, 2023).
Myopati ved behandling av HIV/AIDS: myopati sees ved bruk av nukleosid-analog revers-transkriptase inhibitorer (AZT) og ved protease inhibitorer
Sjekkpunkthemmere: Sjekkpunkthemmere (immunologic check-point inhibitors, ICI) brukes i behandling av kreft i økende grad. Data til nå tyder på at 5-10% får revmatiske symptomer og / eller sykdom relatert til denne kreftbehandlingen. Ledd– og muskelsmerter er vanligst. Artritt oppstår hos 3-4%.
Steroid-myopati: Kan utvikles under steroid-behandling og mistolkes som polymyositt / dermatomyositt eller eksaserbasjon under behandling av av disse. Steroid-myopati ses ved alle typer kortikosteroider, oftest ved triamcinolon, betametason og deksametason. Oftest har pasienten fått langvarig behandling, men akutt steroid-myopati 5-7 dager etter behandlingsstart er også mulig i form av akutt quadriceps myopati og critical illness myopati, oftest ved høydose i.v. Typisk er klinisk forverring under pågående steroid-behandling og ingen effekt av doseøkning. Proksimale muskelgrupper og underekstremiteter rammes, mens nakkemuskler sjelden angripes. Muskelbiopsi viser type II fiberatrofi, sjeldnere type II eller diffus nekrose.
Litteratur:
Doughty CD, 2019 (toksisk myopati)
Brahmer JR, 2018 (Sjekkpunkthemmere: (ASCO guidelines) for behandling av bivirkninger=
Calabrese L, 2018 (Sjekkpunkthemmere)
Minetto MA, 2018 (steroid myopati)
